Vascular anomaly
A vascular anomaly is a kind of birthmark caused by a disorder of the vascular development, although it is not always present at birth. A vascular anomaly is a localized defect in blood vessels that can affect each part of the vasculature (capillaries, arteries, veins, lymphatics or a combination of these). These defects are characterized by an increased number of vessels and vessels that are both enlarged and sinuous. Some vascular anomalies are congenital and therefore present at birth, others appear within weeks to years after birth and others are acquired by trauma or during pregnancy. Inherited vascular anomalies are also described and often present with a number of lesions that increase with patients’ age. Vascular anomalies can also be a part of a syndrome and, occasionally, they can be acquired by trauma. The estimated prevalence of vascular anomalies is 4.5%.[1] Vascular anomalies can occur throughout the whole body (skin, bone, liver, intestines, i.e.), but in 60% of patients vascular anomalies are localized in the head and neck region.[2] Vascular anomalies can present in various ways. Vascular anomalies that are situated deep below the skin, appear blue and are often called cavernous. Superficial vascular anomalies appear as red-coloured stains and are associated with vascular anomalies affecting the dermis. Historically, vascular anomalies have been labeled with descriptive terms, according to the food they resembled (port wine, strawberry, cherry, salmon patch). This imprecise terminology has caused diagnostic confusion, blocked communication and even caused incorrect treatment, as it does not differentiate between various vascular anomalies.[3] However, in 1982, Mulliken introduced a classification that replaced these descriptive terms and gave direction to the management of various vascular anomalies. This classification, based on clinical features, natural history and cellular characteristics, divides vascular anomalies into two groups: vascular tumors and vascular malformations.[4] Although the appearance of both vascular tumors and vascular malformations can resemble, there are important differences between both.
| Correct term | Incorrect terminology commonly used to describe vascular anomalies |
|---|---|
| Hemangioma | Strawberry Hemangioma
Capillary Hemangioma Cavernous Hemangioma |
| Kaposiform hemangioendothelioma | (Capillary) Hemangioma |
| Pyogenic granuloma | Hemangioma |
| Capillary Malformation | Port-wine stain
Capillary Hemangioma |
| Lymphatic Malformation | Lymphangioma
Cystic hygroma |
| Venous Malformation | Cavernous Hemangioma |
| Arteriovenous malformation | Arteriovenous Hemangioma |
Vascular tumors
Vascular tumors, often referred to as hemangiomas, are the most common tumors in infants, occurring in 1-2%. Prevalence is even higher (10%) in premature infants of very low birth weight.[2] Vascular tumors are characterized by overgrowth of normal vessels, which show increased endothelial proliferation. It can be present at birth, but often appears within a couple of weeks after birth or during infancy. There are different kinds of vascular tumors, but the 4 most common types are: infantile hemangioma, congenital hemangioma, kaposiform hemangioendothelioma and pyogenic granuloma.
Infantile Hemangioma
Infantile hemangioma is the most common vascular tumor. It is a benign tumor, which occurs in 4-5% of Caucasian infants, but rarely in dark skinned infants.[5] It occurs in 20% of low weight premature infants and 2.2 to 4.5 times more frequently in females.[6] IH most commonly presents in the head and neck region (60%), but also involves the trunk and extremities. One third of these lesions is present at birth as a telangiectatic stain or ecchymotic area. During the first four weeks of life, 70% to 90% appear. Lesions that are situated beneath the skin may not appear until 3 to 4 months of age, when the tumor is large enough. During the first 9 months, IH undergoes rapid growth, which is faster than the growth of the child. This is called the proliferating phase. After 9 months, the growth of the tumor will decrease and equal the growth of the child for about 3 months. After 12 months, the tumor will start to involute and might even disappear. Involution occurs in one-third of patient by the age of 3 years, in 50% by the age of 5 years and in 72% by the age of 7 years.[7] Involution may result in residual telangiestasis, pallor, atrophy, textural changes and sometimes fibrofatty residuum. Since 90% of IH is small, localized and asymptomatic, treatment mainly consists of observation and awaiting until involution is complete. IH can be treated with corticosteroids, which accelerate involution: in 95% of patients, growth is stabilized and 75% of tumors decrease in size. Intralesional corticosteroids are most effective, but may require additional injections, as the effect is only temporarily. Systemic corticosteroids may cause a number of side-effects and are only used in problematic IH, which is too large to treat with intralesional injections. During the proliferating phase, the tumor is highly vascular. Patients who undergo operative treatment during this period, are at risk for blood loss. Moreover, surgery during this phase, often leads to an inferior aesthetic outcome. However, patients may require intervention during childhood, because 50% of IH leave residual fibrofatty tissue, redundant skin, or damaged structures after involution. Waiting until involution is completed, ensures that the least amount of fibro fatty residuum and excess skin is resected, giving the smallest possible scar.[5] Another option for treatment in the pulsed-dye laser. After involution residual telangiectasias can be treated with laser therapy.
Congenital Hemangioma
Congenital hemangioma can be distinguished from infantile hemangioma because it is fully developed at birth. It forms during prenatal life and has reached its maximal size at birth. Congenital hemangioma can even be diagnosed in utero by prenatal ultrasound. Unlike IH, CH is more common in the extremities, has an equal sex distribution, and is solitary, with an average diameter of 5 cm. It commonly presents in the head and neck and in the lower extremities. Congenital hemangioma are divided into 2 subgroups: the rapidly involuting congenital hemangiomas (RICHs) and the non-involuting congenital hemangiomas(NICHs).
The rapidly involuting congenital hemangioma, RICH, presents at birth as a solitary raised tumor with a central depression, scar, or ulceration surrounded by a rim of pallor. It is noted for its involution, which typically begins several weeks after birth and is completed no later than 14 months of age.[8] After regression RICH may cause a residual deformity, such as atrophic skin and subcutaneous tissue. It mainly affects the limbs (52%), but also the head and neck region (42%) and the trunk (6%).[5]
The non-involuting congenital hemangioma, NICH, presents as a solitary, well-circumscribed reddish-pink to purple plaque with central telangiectasia and hypopigmented rim.[8] In contrast to RICH, NICH does not involute and rarely ulcerates. It persists into late childhood and can even mimic a vascular malformation by growing commensurately with the child. Although NICH can resemble RICH in its external appearance, it can be differentiated from RICH by a greater elevation and coarse telangiectases. It mainly affects the head and neck region (43%), but also the limbs (38%) and the trunk (19%).
Surgical resection for congenital hemangiomas is rarely needed, because RICH undergoes postnatal regression and NICH is benign and often asymptomatic. Resection may be indicated to improve the appearance of the affected area, as long as the surgical scar is less noticeable than the lesion. Other indications are problematic ulcers with persistent bleeding or chronic infection. Although most NICH lesions are non-problematic and do not cause significant deformity, the threshold for resection of NICH is lower, because it neither involutes, nor responds to pharmacotherapy. RICH tumors are observed until involution is completed. Involuted RICH may leave behind atrophic tissue, which can be reconstructed with autologous grafts.[5] It is often best to postpone excision until regression is complete. There are effective pharmacologic treatments, which include intralesional corticosteroid injection, systemic corticosteroid injection, interferon α-2a or α-2b and angiogenic inhibitors. The use of corticosteroids leads to accelerated regression in 30%, stabilization of growth in 40%, lightening of color and softening of the tumor. However, 30% shows minimal or no response. Another drug treatment is interferon α-2a or α-2b. It is often used for patients who did not respond to corticosteroids. Although the response rate is much slower, it has been successful for 80% of children treated.[9] The most serious side effect of interferon is a spastic diplegia. Other therapeutic options are embolization and pulsed-dye laser, which improves residual telangiectasias in RICH and in NICH.
Kaposiform Hemangioendothelioma
Kaposiform hemangioendothelioma (KHE) is a rare vascular neoplasm that is locally aggressive but without metastatic potential. It occurs particularly in the skin, deep soft tissue, retroperitoneum, mediastinum, and rarely in bone. Although lesions occur solitary, they often involve large areas of the body, such as the head/neck region (40%), trunk (30%), or extremity (30%). Usually, it is present at birth as a flat, reddish-purple, tense and edematous lesion. Although half of lesions are congenital, 58% of KHE develop during infancy, 32% between age 1 and 10 years (32%) and 10% after 11 years of age. Moreover, adult onset has been described too with mainly males being affected. Both sexes are affected equally in children. Lesions are often greater than 5 cm in diameter and can cause visible deformity and pain. During early childhood, KHE may enlarge and after 2 years of age, it may partially regress. Though, it usually persists longterm. In addition, 50% of patients suffer from coagulopathy due to thrombocytopenia (<25,000/mm3), presenting with petechiae and bleeding. This is called the Kasabach-Merritt Phenomenon, which is caused by trapping of platelets and other clotting factors within the tumor. Kasabach-Merritt Phenomenon is less likely in patients with lesions less than 8 cm. As two-thirds of adult-onset KHE tumors are less than 2 cm, KHE in adults is rarely associated with Kasabach-Merritt Phenomenon.[5] Patients with KHE and Kasabach-Merritt Phenomenon present with petechiae and ecchymosis. Most KHE tumors are diffuse involving multiple tissue planes and important structures. Resection of KHE is thus often difficult. Treatment of kaposiform hemangioendothelioma is therefore medical. The primary drug is interferon alfa, which is successful in 50% of children.[9] Another option is vincristine, which has lots of side-effects, but has a response rate of 90%. Drug therapy is often used in shrinking the tumor and treating the coagulopathy. However, many of these kaposiform hemangioendotheliomas do not completely regress and remain as a much smaller asymptomatic tumor. However, KHE still has a high mortality rate of 30%. Although complete surgical removal with a large margin has the best reported outcome, it is usually not done because of the risk of bleeding, extensiveness, and the anatomic site of the lesion.[10] Operative management may be possible for small or localized lesions. Removal of larger areas also may be indicated for symptomatic patients or for patients who have failed farmacotherapy. Resection is not required for lesions that are not causing functional problems, because KHE is benign and because resection could cause deformity.
Pyogenic granuloma

Pyogenic granuloma, also known as lobular capillary hemangioma, is a small benign vascular tumor that primarily involves the skin (88.2%) and mucous membranes.[5] Pyogenic granuloma appears as a red macule that grows rapidly, turns into a papule and eventually becomes pedunculated, being attached to a narrow stalk.[7] The average diameter of these lesions is 6.5 mm.[5] Although these lesions are small, they are often complicated by bleeding, crusting and ulceration. Microscopically, pyogenic granulomas are characterized by vascular proliferation amidst granulation tissue and chronic inflammatory infiltrate.[11]
Pyogenic granulomas are rarely congenital. It commonly develops in infants: 42.1% develops within the first 5 years of life.[5] This vascular tumor is twice as common in males as in females and 25% of lesions seem to be associated with trauma, an underlying cutaneous condition, pregnancy, hormonal alterations and medications.[11] Pyogenic granulomas can also arise within a capillary malformation. Of all pyogenic granulomas, 62% is distributed on the head or neck, occurring mainly on the cheek and in the oral cavity. Lesions on the face may cause visible deformity.
Numerous treament methods have been described for pyogenic granuloma. Lesions involving the reticular dermis, may be out of the reach of pulsed-dye laser, cautery or shave excision and therefore have a recurrence rate of 43.5%.[5] Definitive management requires full-thickness skin excision. Other options are currettage or laser therapy. Furthermore, thorough currettage and cauterization are often used for small lesions and full-thickness excision for larger lesion.
Vascular malformations
Vascular malformation is a collective term for different disorders of the vasculature (errors in vascular development). It can be a disorder of the capillaries, arteries, veins and lymphatic vessels or a disorder of a combination of these (lesions are named based on the primary vessel that is malformed). A vascular malformation consists of a cluster of deformed vessels, due to an error in vascular development (dysmorphogenesis). However, endothelial turnover is stable in these defects. Congenital vascular malformations are always already present at birth, although they are not always visible. In contrast to vascular tumors, vascular malformations do not have a growth phase, nor an involution phase. Vascular malformations tend to grow proportionately with the child.[12] Vascular malformations never regress, but persist throughout life. Vascular malformations can be divided into slow-flow, fast-flow and complex-combined types.[13]
Slow-flow vascular malformations
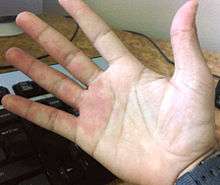
- Capillary malformation (also known as port-wine stains): Capillary malformations are flat, reddish lesions that typically affect the skin, mostly around the head and the neck, and which darken with age, in contrast to birthmarks such as salmon patch, Nevus simplex or vascular stain, which lighten or disappear within the first few years of life. Capillary malformations constitute 11% of the vascular malformations.[1] Syndromes associated with capillary malformations are: Sturge-Weber syndrome and Klippel-Trenaunay syndrome.[13] Capillary malformations can be treated with IPL-(Intensed-pulsed-light)-therapy or surgical reduction.[2]
- Venous malformation is a bluish lesion compressible on palpation; the masses enlarge with physical activity or if in a dependent position. The bluish lesion is caused by dilated venous vessels. Venous malformations can be painful in the morning due to stasis and microthrombi within the veins. Venous malformations usually occur in the head and neck.[12] Venous malformations are the most common vascular anomaly, making up 40% of all vascular malformations.[1] They can be treated with sclerotherapy and surgical reduction[2]

- Lymphatic malformation is a benign growth of the lymphatic system.[14] They result from a blockage or defect of the lymphatic vessels as they are forming. 28% of all vascular malformations are lymphatic malformations.[1] Lymphatic malformations can be treated with sclerotherapy and surgical reduction.[2]
Fast flow vascular malformations
All fast-flow malformations are malformations involving arteries. They constitute about 14% of all vascular malformations.[1]
- Arterial malformation
- Arteriovenous fistula (AVF) : a lesion with a direct communication via fistulae between an artery and a vein.[2]
- Arteriovenous malformation : a lesion with a direct connection between an artery and a vein, without an intervening capillary bed, but with an interposed nidus of dysplastic vascular channels in between.[12]
Combined-complex vascular malformations
a combination of various vascular malformations. They are 'complex' because they involve a combination of two different types of vessels.
- CVM: capillary venous malformation
- CLM: capillary lymphatic malformation
- LVM: lymphatic venous malformation
- CLVM: capillary lymphatic venous malformation. CLVM is associated with Klippel-Trenaunay syndrome
- AVM-LM: Arteriovenous malformation- lymphatic malformation
- CM-AVM: capillary malformation- arteriovenous malformation[13]
References
- 1 2 3 4 5 6 Greene, AK (January 2011). "Vascular anomalies: current overview of the field.". Clinics in plastic surgery. 38 (1): 1–5. PMID 21095467. doi:10.1016/j.cps.2010.08.004.
- 1 2 3 4 5 6 Ernemann, U; Kramer, U; Miller, S; Bisdas, S; Rebmann, H; Breuninger, H; Zwick, C; Hoffmann, J (July 2010). "Current concepts in the classification, diagnosis and treatment of vascular anomalies.". European journal of radiology. 75 (1): 2–11. PMID 20466500. doi:10.1016/j.ejrad.2010.04.009.
- ↑ Hassanein, AH; Mulliken, JB; Fishman, SJ; Greene, AK (January 2011). "Evaluation of terminology for vascular anomalies in current literature.". Plastic and Reconstructive Surgery. 127 (1): 347–51. PMID 21200229. doi:10.1097/PRS.0b013e3181f95b83.
- ↑ Mulliken, JB; Glowacki, J (March 1982). "Hemangiomas and vascular malformations in infants and children: a classification based on endothelial characteristics.". Plastic and Reconstructive Surgery. 69 (3): 412–22. PMID 7063565. doi:10.1097/00006534-198203000-00002.
- 1 2 3 4 5 6 7 8 9 Greene, AK (January 2011). "Management of hemangiomas and other vascular tumors.". Clinics in plastic surgery. 38 (1): 45–63. PMID 21095471. doi:10.1016/j.cps.2010.08.001.
- ↑ Kim, LH; Hogeling, M; Wargon, O; Jiwane, A; Adams, S (April 2011). "Propranolol: useful therapeutic agent for the treatment of ulcerated infantile hemangiomas.". Journal of pediatric surgery. 46 (4): 759–63. PMID 21496551. doi:10.1016/j.jpedsurg.2011.01.012.
- 1 2 Van Aalst, JA; Bhuller, A; Sadove, AM (July 2003). "Pediatric vascular lesions.". The Journal of craniofacial surgery. 14 (4): 566–83. PMID 12867875. doi:10.1097/00001665-200307000-00032.
- 1 2 Beck, DO; Gosain, AK (June 2009). "The presentation and management of hemangiomas.". Plastic and Reconstructive Surgery. 123 (6): 181e–91e. PMID 19483535. doi:10.1097/PRS.0b013e3181a65c59.
- 1 2 Hentz, Ed. Vincent R. (2006). "209: Vascular Anomalies of the Upper Extremity". The hand and upper limb ; part 2 (2. ed.). Philadelphia, Pa.: Saunders Elsevier. ISBN 0-7216-8819-5.
- ↑ Hermans DJ, van Beynum IM, van der Vijver RJ, Kool LJ, de Blaauw I, van der Vleuten CJ (May 2011). "Kaposiform hemangioendothelioma with Kasabach-Merritt syndrome: a new indication for propranolol treatment.". Journal of pediatric hematology/oncology. 33 (4): e171–3. PMID 21516018. doi:10.1097/MPH.0b013e3182152e4e.
- 1 2 Gupta, A; Kozakewich, H (January 2011). "Histopathology of vascular anomalies.". Clinics in plastic surgery. 38 (1): 31–44. PMID 21095470. doi:10.1016/j.cps.2010.08.007.
- 1 2 3 Chim, H; Drolet, B; Duffy, K; Koshima, I; Gosain, AK (August 2010). "Vascular anomalies and lymphedema.". Plastic and Reconstructive Surgery. 126 (2): 55e–69e. PMID 20679788. doi:10.1097/PRS.0b013e3181df803d.
- 1 2 3 Enjolras, O (2007). "Introduction: ISSVA Classification". Color atlas of vascular tumors and vascular malformations. Cambridge [u.a.]: Cambridge University Press. ISBN 978-0-521-84851-0.
- ↑ Perkins JA, Manning SC, Tempero RM, Cunningham MJ, Edmonds JL, Hoffer FA, Egbert MA (June 2010). "Lymphatic malformations: current cellular and clinical investigations.". Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 142 (6): 789–94. PMID 20493347. doi:10.1016/j.otohns.2010.02.025.