Ultrafast laser spectroscopy
Ultrafast laser spectroscopy is a spectroscopic technique that uses ultrashort pulse lasers for the study of dynamics on extremely short time scales (attoseconds to nanoseconds). Different methods are used to examine dynamics of charge carriers, atoms and molecules. Many different procedures have been developed spanning different time scales and photon energy ranges; some common methods are listed below.
Attosecond-to-picosecond spectroscopy
Dynamics on the as to fs time scale is in general too fast to be measured electronically. Most measurements are done by employing a sequence of ultrashort light pulses to initiate a process and record its dynamics. The width of the light pulses have to be on the same scale as the dynamics that is to be measured.
Light sources
Titanium-sapphire laser
Ti-sapphire lasers are tunable lasers which emit red and near-infrared light (700 nm- 1100 nm).Ti-sapphire laser systems use Ti-sapphire as a gain medium. The pulses will go into a stretcher where the pulse duration is stretched, and then to a regenerate amplifier, where the pulse energy is amplified. The output pulses from the regenerate amplifier are further sent to a multi-pass amplifier, where the pulses can be amplified to even higher energies. The pulses from either the regenerate amplifier or the multi-pass amplifier are sent to a compressor, where the pulse duration is compressed.
Dye laser
A dye laser is a four-level laser which uses organic dye as the gain medium. Pumped by a laser with a fixed wavelength, due to various dye types you use, different dye lasers can emit beams with different wavelengths. A ring laser design is most often used in a dye laser system. Also, tuning elements, such as a diffraction grating or prism, are usually incorporated in the cavity. This allows only light in a very narrow frequency range to resonate in the cavity and be emitted as laser emission. The wide tuneability range, high output power, and pulsed or CW operation make the dye laser particularly useful in many physical & chemical studies.
Fiber laser
A fiber laser is usually generated first from a laser diode. The laser diode then couples the light into a fiber where it will be confined. Different wavelengths can be achieved with the use of doped fiber. The pump light from the laser diode will excite a state in the doped fiber which can then drop in energy causing a specific wavelength to be emitted. This wavelength may be different from that of the pump light and more useful for a particular experiment.
X-ray generation
Ultrafast optical pulses can be used to generate x-ray pulses in multiple ways. An optical pulse can excite an electron pulse via the photoelectric effect, and acceleration across a high potential gives the electrons kinetic energy. When the electrons hit a target they generate both characteristic x-rays and bremsstrahlung. A second method is via laser induced plasma. When very high intensity laser light is incident on a target, it strips electrons off the target creating a negatively charged plasma cloud. The strong Coulomb force due to the ionized material in the center of the cloud quickly accelerates the electrons back to towards the nuclei left behind. Upon collision with the nuclei, Bremsstrahlung and characteristic emission x-rays are given off. This method of x-ray generation scatters photons in all directions, but also generates picosecond x-ray pulses.
Conversion and characterization
Pulse characterization
In order for accurate spectroscopic measurements to be made, several characteristics of the laser pulse need to be known; pulse duration, pulse energy, spectral phase and spectral shape are among some of these.[1] Information about pulse duration can be determined through autocorrelation measurements, or from cross correlation with another well characterized pulse. Methods allowing for complete characterization of pulses include frequency-resolved optical gating (FROG) and spectral phase interferometry for direct electric-field reconstruction (SPIDER).
Pulse shaping
Pulse shaping is to modify the pulses from the source in a well-defined manner, including manipulation on pulse’s amplitude, phase and duration. To amplify pulse’s intensity, chirped pulse amplification is generally applied, which includes a pulse stretcher, amplifier and compressor. It will not change the duration or phase of the pulse during the amplification. Pulse compression (shorten the pulse duration) is achieved by first chirping the pulse in a nonlinear material and broadening the spectrum, with a following compressor for chirp compensation. Fiber compressor is generally used in this case. Pulse shapers usually refer to optical modulators which applies Fourier transforms to laser beam. Depending on which property of light is controlled, modulators are called intensity modulators, phase modulators, polarization modulators, spatial light modulators. Depending on the modulation mechanism, optical modulators are divided into Acoustic-optic modulators, Electro-optic modulators, Liquid crystal modulators etc. Each is dedicated into different applications.[2]
High harmonic generation
High harmonic generation (HHG) is the nonlinear process where intense laser radiation is converted from one fixed frequency to high harmonics of that frequency by ionization and recollision of an electron. It was first observed in 1987 by McPherson et al. who successfully generated harmonic emission up to the 17th order at 248 nm in neon gas.[3] HHG is seen by focusing an ultra-fast, high-intensity, near-IR pulse into a noble gas at intensities of (1013–1014 W/cm2) and it generates coherent pulses in the XUV to Soft X-ray (100–1 nm) region of the spectrum. It is realizable on a laboratory scale (table-top systems) as opposed to large free electron-laser facilities.
High harmonic generation in atoms is well understood in terms of the three-step model (ionization, propagation, and recombination). Ionization: The intense laser field modifies the Coulomb potential of the atom, electron tunnels through the barrier and ionize. Propagation: The free electron accelerates in the laser field and gains momentum. Recombination: When the field reverses, the electron is accelerated back toward the ionic parent and releases a photon with very high energy.[4]
Frequency conversion techniques
Different spectroscopy experiments require different excitation or probe wavelengths. For this reason frequency conversion techniques are commonly used to extend the operational spectrum of existing laser light sources. The most widespread conversion techniques rely on using crystals with second order non linearity to perform either parametric amplification or frequency mixing. Frequency mixing works by superimposing two beams of equal or different wavelengths to generate a signal which is a higher harmonic or the sum frequency of the first two. Parametric amplification overlaps a weak probe beam with a higher energy pump beam in a non linear crystal such that the weak beam gets amplified and the remaining energy goes out as a new beam called the idler. This approach has the capability of generating output pulses that are shorter than the input ones. Different schemes of this approach have been implemented. Examples are: optical parametric oscillator (OPO), optical parametric amplifier (OPA), non-collinear parametric amplifier (NOPA).
Techniques
Ultra-fast transient absorption
This method is typical of 'pulse-probe' experiments, where a pulsed laser is used to excite a molecule's electrons from their ground states to higher-energy excited states. A probing light source, typically a xenon arc lamp, is used to obtain an absorption spectrum of the compound at various times following its excitation. As the excited molecules absorb the probe light, they are further excited to even higher states. After passing through the sample, the unabsorbed light from the arc lamp continues to an avalanche photodiode array, and the data is processed to generate an absorption spectrum of the excited state. Since all the molecules in the sample will not undergo the same dynamics simultaneously, this experiment must be carried out many times, and the data must be averaged in order to generate spectrums with accurate intensities and peaks. Unlike TCSPC, this technique can be carried out on non-fluorescent samples.
Ultrafast transient absorption can use almost any probe light, so long as the probe is of a pertinent wavelength or set of wavelengths. A monochromator and photomultiplier tube in place of the avalanche photodiode array, allows observation of a single probe wavelength, and thus allows probing of the decay kinetics of the excited species. The purpose of this setup is to take kinetic measurements of species that are otherwise nonradiative, and specifically it is useful for observing species that have short-lived and non-phosphorescent populations within the triplet manifold as part of their decay path. It should be noted that the pulsed laser in this setup is used both as a primary excitation source, and a clock signal for the ultrafast measurements. Although laborious and time-consuming, the monochromator position may also be shifted to allow absorbance decay profiles to be constructed, ultimately to the same effect as the above method.
Time-resolved photoelectron spectroscopy and two-photon photoelectron spectroscopy
Time-resolved photo-electron spectroscopy and two-photon photoelectron spectroscopy (2PPE) combine a pump-probe scheme with angle-resolved photoemission. A first laser pulse is used to excite a material, a second laser pulse ionizes the system. The kinetic energy of the electrons from this process are then detected, through various methods including energy mapping, time of flight measurements etc. As above, the process is repeated many times, with different time delays between the probe pulse and the pump pulse. This builds up a picture of how the molecule relaxes over time. A variation of this method looks at the positive ions created in this process, and is called time-resolved photo-ion spectroscopy (TRPIS)
Multidimensional spectroscopy
Using the same principles pioneered by 2D-NMR experiments, multidimensional optical spectroscopy is possible using ultrafast pulses. Different frequencies can probe various dynamic molecular processes to differentiate between inhomogeneous and homogeneous line broadening as well as identify coupling between the measured spectroscopic transitions. If two oscillators are coupled together, be it intramolecular vibrations or intermolecular electronic coupling, the added dimensionality will resolve anharmonic responses not identifiable in linear spectra. A typical 2D pulse sequence consists of an initial pulse to pump the system into coherent superposition of states, followed by a phase conjugate second pulse that pushes the system into a non-oscillating excited state, and finally, a third pulse that converts back to a coherent state that produces a measurable pulse.[5] A 2D frequency spectrum can then be recorded by plotting the Fourier transform of the delay between the first and second pulses on one axis, and the Fourier transform of the delay between a detection pulse relative to the signal-producing third pulse on the other axis. 2D spectroscopy is an example of a four wave mixing experiment, and the wavevector of the signal will be the sum of the three incident wavevectors used in the pulse sequence. Multidimensional spectroscopy exist in infrared and visible variants as well as combinations using different wavelength regions.
Ultrafast imaging
Most ultrafast imaging techniques are variations on standard pump-probe experiments. Some commonly used techniques are Electron Diffraction imaging,[6] Kerr Gated Microscopy,[7] imaging with ultrafast electron pulses [8] and terahertz imaging.[9] Novel applications of these imaging techniques are constantly arising. This is particularly true in the biomedical community where safe and non-invasive techniques for diagnosis are always of interest. Terahertz imaging has recently been used to identify areas of decay in tooth enamel and image the layers of the skin. Additionally it has shown to be able to successfully distinguish a region of breast carcinoma from healthy tissue.[9] Another technique called Serial Time-encoded amplified microscopy has shown to have the capability of even earlier detection of trace amount of cancer cells in the blood.[10] Other non-biomedical applications include ultrafast imaging around corners or through opaque objects.
Femtosecond up-conversion
Femtosecond up-conversion is a pump-probe technique that uses nonlinear optics to combine the fluorescence signal and probe signal to create a signal with a new frequency via photon upconversion, which is subsequently detected. The probe scans through delay times after the pump excites the sample, generating a plot of intensity over time.[11]
Applications
Applications of femtosecond spectroscopy to biochemistry
Ultrafast processes are found throughout biology. Until the advent of femtosecond methods, many of the mechanism of such processes were unknown.[12][13] Examples of these include the cis-trans photoisomerization of the rhodopsin chromophoreretinal, excited state and population dynamics of DNA, and the charge transfer processes in photosynthetic reaction centers[13] Charge transfer dynamics in photosynthetic reaction centers has a direct bearing on man’s ability to develop light harvesting technology, while the excited state dynamics of DNA has implications in diseases such as skin cancer.[14][15] Advances in femtosecond methods are crucial to the understanding of ultrafast phenomena in nature.
Photodissociation and femtosecond probing
Photodissociation is a chemical reaction in which a chemical compound is broken down by photons. It is defined as the interaction of one or more photons with one target molecule. Any photon with sufficient energy can affect the chemical bonds of a chemical compound, such as visible light, ultraviolet light, x-rays and gamma rays. The technique of probing chemical reactions has been successfully applied to unimolecular dissociations. The possibility of using a femtosecond technique to study bimolecular reactions at the individual collision level is complicated by the difficulties of spatial and temporal synchronization. One way to overcome this problem is through the use of Van der Waals complexes of weakly bound molecular cluster. Femtosecond techniques are not limited to the observation of the chemical reactions, but can even exploited to influence the course of the reaction. This can open new relaxation channels or increase the yield of certain reaction products.
Picosecond-to-nanosecond spectroscopy
Streak camera
Unlike attosecond and femtosecond pulses, the duration of pulses on the nanosecond timescale are slow enough to be measured through electronic means. Streak cameras translate the temporal profile of pulses into that of a spatial profile; that is, photons that arrive on the detector at different times arrive at different locations on the detector.
Time-correlated single photon counting
Time-correlated single photon counting (TCSPC) is used to analyze the relaxation of molecules from an excited state to a lower energy state. Since various molecules in a sample will emit photons at different times following their simultaneous excitation, the decay must be thought of as having a certain rate rather than occurring at a specific time after excitation. By observing how long individual molecules take to emit their photons, and then combining all these data points, an intensity vs. time graph can be generated that displays the exponential decay curve typical to these processes. However, it is difficult to simultaneously monitor multiple molecules. Instead, individual excitation-relaxation events are recorded and then averaged to generate the curve.
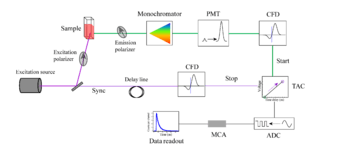
This technique analyzes the time difference between the excitation of the sample molecule and the release of energy as another photon. Repeating this process many times will give a decay profile. Pulsed lasers or LEDs can be used as a source of excitation. Part of the light passes through the sample, the other to the electronics as "sync" signal. The light emitted by the sample molecule is passed through a monochromator to select a specific wavelength. The light then is detected and amplified by a photomultiplier tube (PMT). The emitted light signal as well as reference light signal is processed through a constant fraction discriminator (CFD) which eliminates timing jitter. After passing through the CFD, the reference pulse activates a time-to-amplitude converter (TAC) circuit. The TAC charges a capacitor which will hold the signal until the next electrical pulse. In reverse TAC mode the signal of "sync" stops the TAC. This data is then further processed by an analog to digital converter (ADC) and multi-channel analyzer (MCA) to get a data output. To make sure that the decay is not biased to early arriving photons, the photon count rate is kept low (usually less than 1% of excitation rate).[16]
This electrical pulse comes after the second laser pulse excites the molecule to a higher energy state, and a photon is eventually emitted from a single molecule upon returning to its original state. Thus, the longer a molecule takes to emit a photon, the higher the voltage of the resulting pulse. The central concept of this technique is that only a single photon is needed to discharge the capacitor. Thus, this experiment must be repeated many times to gather the full range of delays between excitation and emission of a photon. After each trial, a pre-calibrated computer converts the voltage sent out by the TAC into a time and records the event in a histogram of time since excitation. Since the probability that no molecule will have relaxed decreases with time, a decay curve emerges that can then be analyzed to find out the decay rate of the event.[17]
A major complicating factor is that many decay processes involve multiple energy states, and thus multiple rate constants. Though non-linear least squared analysis can usually detect the different rate constants, determining the processes involved is often very difficult and requires the combination of multiple ultra-fast techniques. Even more complicating is the presence of inter-system crossing and other non-radiative processes in a molecule. A limiting factor of this technique is that it is limited to studying energy states that result in fluorescent decay. The technique can also be used to study relaxation of electrons from the conduction band to the valence band in semiconductors.[18]
See also
- Time-resolved spectroscopy
- Terahertz time-domain spectroscopy (THz-TDS)
- Electronic configuration
- Atomic spectral line
References
- ↑ Dr. Rüdiger Paschotta (12 August 2015). "Encyclopedia of Laser Physics and Technology - pulse characterization, optical, pulse duration, spectral phase, pulses, FROG, SPIDER".
- ↑ Dr. Rüdiger Paschotta (22 March 2013). "Encyclopedia of Laser Physics and Technology - optical modulators, acousto-optic, electro-optic".
- ↑ B.S, Wagner (2001). High-Order Harmonic Generation from Molecules. Case Western Reserve University.
- ↑ Dinh, Khuong (2012). Phase-Matched High Order Harmonic Generation and Applications. Swinburne University of Technology Melbourne.
- ↑ [ Mukamel, S. Annu. Rev. Phys. Chem. 2000, 51, 691-729.]
- ↑ C. D. LIN* AND JUNLIANG XU, PHYS. CHEM. CHEM. PHYS., 2012, 14, 13133–13145
- ↑ GUNDLACH L., PIOTROWIAK P, OPT. LETT. 33 2008, 992
- ↑ HENSLEY C., YANG J., CENTURION M., PHYS. RE V. LETT., 2012, 109, 133202-1-133202-5,
- 1 2 PICKWELL E., WALLACE V., J. PHYS. D: APPL. PHYS.,2012, 39, R301-R310
- ↑ Goda K. et al., PNAS 2012, 109, 11630-11635
- ↑ http://www.dmphotonics.com/Femtosecond%20Fluorescene%20Up-Conversion%20Spectrometer%20with%20femtosecond%20Ti%20sapphire%20laser/Trotzky_JPhysDApplPhys_42_2009.pdf
- ↑ [ Mathies, R. A. In Ultrafast Processes in Chemistry and Photobiology; El-Sayed, M.A.; Tanaka, I.; Molin, Y.; Ed. Oxford: Cambridge, 1995; pp 215-225.]
- 1 2 [ Sundström, V. Annu.Rev.Phys.Chem 2008, 59, 53-77.]
- ↑ [Schlau-Cohen, G., S.; De Re, E.; Cogdell, R. J. ; Flemming, G. R.; J. Phys. Chem. Lett. 2013. 3, 2487-2492]
- ↑ [ Martinez, T.J.; Hudock, H.R. ChemPhysChem. 2008, 9, 2486-2490]
- ↑ http://www.picoquant.com/images/uploads/page/files/7253/technote_tcspc.pdf
- ↑ Lakowicz, Joseph R. (2006). Principles of fluorescence spectroscopy. Berlin: Springer. ISBN 0-387-31278-1.
- ↑ "Characterization of semiconductor devices and wafer materials via sub-nanosecond time-correlated single-photon counting".
External links
- Ultrafast studies of single semiconductor and metal nanostructures through transient absorption microscopy, a Chemical Science mini review by Gregory Hartland
- W. Becker: The bh TCSPC Handbook., Fifth Edition, 2012, (Becker & Hickl GmbH, PDF file, 77 MB)
- Ultrafast Lasers: An animated guide to the functioning of Ti:Sapphire lasers and amplifiers.