Trypanosoma brucei
| Trypanosoma brucei | |
|---|---|
 | |
| Trypanosoma brucei brucei TREU667 (Bloodstream form, phase contrast picture. Black bar indicates 10 µm.) | |
| Scientific classification | |
| (unranked): | Excavata |
| Phylum: | Euglenozoa |
| Class: | Kinetoplastea |
| Order: | Trypanosomatida |
| Genus: | Trypanosoma |
| Species: | T. brucei |
| Binomial name | |
| Trypanosoma brucei Plimmer & Bradford, 1899 | |
| Subspecies | |
|
T. b. brucei | |

Trypanosoma brucei is a species of parasitic kinetoplastid belonging to the genus Trypanosoma. The parasite is the cause of a vector-borne disease of vertebrate animals, including humans, carried by genera of tsetse fly in sub-Saharan Africa. In humans T. brucei causes African trypanosomiasis, or sleeping sickness. In animals it causes animal trypanosomiasis, also called nagana in cattle and horses. T. brucei has traditionally been grouped into three subspecies: T. b. brucei, T. b. gambiense and T. b. rhodesiense.[1] The first is a parasite of non-human vertebrates, while the latter two are the known parasites of humans. Only rarely can the T. b. brucei infect a human.[2]
T. brucei is transmitted between mammal hosts by an insect vector belonging to different species of tsetse fly (Glossina). Transmission occurs by biting during the insect's blood meal. The parasites undergo complex morphological changes as they move between insect and mammal over the course of their life cycle. The mammalian bloodstream forms are notable for their cell surface proteins, variant surface glycoproteins, which undergo remarkable antigenic variation, enabling persistent evasion of host adaptive immunity leading to chronic infection. T. brucei is one of only a few pathogens known to cross the blood brain barrier.[3] There is an urgent need for the development of new drug therapies, as current treatments can have severe side effects and can prove fatal to the patient.[4]
Whilst not historically regarded as T. brucei subspecies due to their different means of transmission, clinical presentation, and loss of kinetoplast DNA, genetic analyses reveal that T. equiperdum and T. evansi are evolved from parasites very similar to T. b. brucei, and are thought to be members of the brucei clade.[5]
The parasite was discovered in 1894 by Sir David Bruce, after whom the scientific name was given in 1899.[6][7]
Species
T. brucei comprises a species complex that includes:
- T. brucei gambiense — Causes slow onset chronic trypanosomiasis in humans. Most common in central and western Africa, where humans are thought to be the primary reservoir.[8]
- T. brucei rhodesiense — Causes fast onset acute trypanosomiasis in humans. Most common in southern and eastern Africa, where game animals and livestock are thought to be the primary reservoir.[8]
- T. brucei brucei — Causes animal trypanosomiasis, along with several other species of Trypanosoma. T. b. brucei is not infective to humans due to its susceptibility to lysis by trypanosome lytic factor-1 (TLF-1).[9] However, it is closely related to, and shares fundamental features with the human-infective subspecies.
Structure
T. brucei is a typical unicellular eukaryotic cell, and measures 8 to 50 μm in length. It has an elongated body having a streamlined and tapered shape. Its cell membrane (called pellicle) encloses the cell organelles, including the nucleus, mitochondria, endoplasmic reticulum, Golgi apparatus, and ribosomes. In addition, there is an unusual organelle called the kinetoplast, which is made up of numerous circular DNA (mitochondrial DNA) and functions as a single large mitochondrion. The kinetoplast lies near the basal body with which it is indistinguishable under microscope. From the basal body arises a single flagellum that run towards the anterior end. Along the body surface, the flagellum is attached to the cell membrane forming an undulating membrane. Only the tip of the flagellum is free at the anterior end.[10] The cell surface of the bloodstream form features a dense coat of variant surface glycoproteins (VSGs) which is replaced by an equally dense coat of procyclins when the parasite differentiates into the procylic in the tsetse fly midgut.[11]

Trypanosomatids show several different classes of cellular organisation of which two are adopted by Trypanosoma brucei at different stages of the life cycle:[10]
- Epimastigote, which is found in tsetse fly. Its kinetoplast and basal body lie anterior to the nucleus, with a long flagellum attached along the cell body. The flagellum starts from the centre of the body.
- Trypomastigote, which is found in mammalian hosts. The kinetoplast and basal body are posterior of nucleus. The flagellum arises from the posterior end of the body.
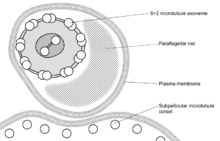
These names are derived from the Greek mastig- meaning whip, referring to the trypanosome's whip-like flagellum. The trypanosome flagellum has two main structures. It is made up of a typical flagellar axoneme which lies parallel to the paraflagellar rod, a lattice structure of proteins unique to the kinetoplastida, euglenoids and dinoflagellates.
The microtubules of the flagellar axoneme lie in the normal 9+2 arrangement, orientated with the + at the anterior end and the - in the basal body. The a cytoskeletal structure extends from the basal body to the kinetoplast. The flagellum is bound to the cytoskeleton of the main cell body by four specialised microtubules, which run parallel and in the same direction to the flagellar tubulin.
The flagellar function is twofold — locomotion via oscillations along the attached flagellum and cell body, and attachment to the fly gut during the procyclic phase.[12]
Life cycle

T. brucei completes its life cycle between tsetsefly (of the genus Glossina) and mammalian hosts, including humans, cattle, horses, and wild animals.
In mammalian host
Infection occurs when a vector tsetse fly bites a mammalian host. The fly injects the metacyclic trypomastigotes into the skin tissue. The trypomastigotes enter the lymphatic system and into the bloodstream. The initial trypomastigotes are short and stumpy. Once inside the bloodstream, they grow into long and slender forms. Then, they multiply by binary fission. The daughter cells then become short and stumpy again.[13][14] The long slender forms are able to penetrate the blood vessel endothelium and invade extravascular tissues, including the central nervous system (CNS).[12]
Sometimes, wild animals can be infected by the tsetse fly and they act as reservoirs. In these animals, they do not produce the disease, but the live parasite can be transmitted back to the normal hosts.[13]
In tsetse fly
The short and stumpy trypomastigotes are taken up by tsetse fly during blood meal. The trypomastigotes enter the midgut of the fly where they become procyclic trypomastigotes. These rapidly divide to become epimastigotes. The epimastigotes migrate from the gut via the proventriculus to the salivary glands where they get attached to the salivary gland epithelium. In the salivary glands, some parasites detach and undergo transformation into short and stumpy trypomastigotes. These become the infective metacyclic trypomastigotes. They are injected into the mammalian host along with the saliva on biting. Complete development in the fly takes about 20 days.[13][14]
Reproduction
Binary fission
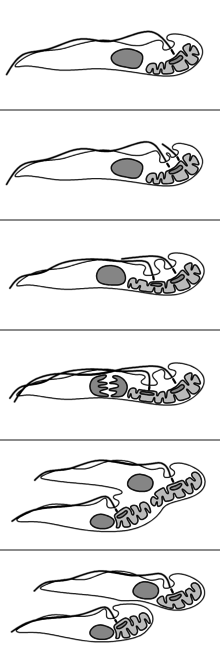
The reproduction of T. brucei is unusual compared to most eukaryotes. The nuclear membrane remains intact and the chromosomes do not condense during mitosis. The basal body, unlike the centrosome of most eukaryotic cells, does not play a role in the organisation of the spindle and instead is involved in division of the kinetoplast. The events of reproduction are:[10]
- The basal body duplicates and both remain associated with the kinetoplast. Each basal body forms a separate flagellum.
- Kinetoplast DNA undergoes synthesis then the kinetoplast divides coupled with separation of the two basal bodies.
- Nuclear DNA undergoes synthesis while a new flagellum extends from the younger, more posterior, basal body.
- The nucleus undergoes mitosis.
- Cytokinesis progresses from the anterior to posterior.
- Division completes with abscission.
Meiosis
In the 1980s, DNA analyses of the developmental staged of T. brucei started to indicate the trypomastigote in tsetse fly undergo meiosis, i.e. sexual reproduction stage.[15] But it is not always necessary for a complete life cycle.[16] The existence of meiosis-specific proteins was reported in 2011.[17] The haploid gametes (daughter cells produced after meiosis) were discovered in 2014. The haploid trypomastigote-like gametes can interact with each other via their flagella and undergo cell fusion (the process called syngamy).[18][19] Thus, in addition to binary fission, T. brucei can multiply by sexual reproduction. Trypanosomes belong to the supergroup Excavata and are one of the earliest diverging lineages among eukaryotes.[20] The discovery of sexual reproduction in T. brucei supports the hypothesis that meiosis and sexual reproduction are ancestral and ubiquitous features of eukaryotes.[21]
Infection and pathogenicity
The insect vectors for T. brucei are different species of tsetse fly (genus Glossina). The major vectors of T. b. gambiense, causing West African sleeping sickness, are G. palpapalis, G. tachinoides, and G. fuscipes. While the principal vectors of T. b. rhodesiense, causing East African sleeping sickness, are G. morsitans, G. pallidipes, and G. Swynnertoni. Animal trypanosomiasis is transmitted by a dozen species of Glossina.[22]
In later stages of a T. brucei infection of a mammalian host the parasite may migrate from the bloodstream to also infect the lymph and cerebrospinal fluids. It is under this tissue invasion that the parasites produce the sleeping sickness.[13]
In addition to the major form of transmission via the tsetse fly T. brucei, may be transferred between mammals via bodily fluid exchange, such as by blood transfusion or sexual contact, although this is thought to be rare.[23][24]
Distribution
T. brucei is found where the tsetse fly vectors are prevalent. It is present in tropical and subtropical areas of Africa north of the equator, covering East, Central and West Africa.[10] Hence, the equatorial region of Africa is called the "sleeping sickness" belt. However, the specific type of the trypanosome differs according to geography. T. b. rhodesiense is found primarily in East Africa (Botswana, Ethiopia, Kenya, Malawi, Tanzania, Uganda, Zaire, and Zimbabwe), while T. b. gambiense is found in Central and West Africa.[11]
Evolution
Trypanosoma brucei gambiense evolved from a single progenitor ~10,000 years ago.[25] It is evolving asexually and its genome shows the Meselson effect.
Genetics
There are two subpopulations of T. b. gambiense that possesses two distinct groups that differ in genotype and phenotype. Group 2 is more akin to T. b. brucei than group 1 T. b. gambiense.[26]
All T. b. gambiense are resistant to killing by a serum component — trypanosome lytic factor (TLF) of which there are two types: TLF-1 and TLF-2. Group 1 T. b. gambiense parasites avoid uptake of the TLF particles while those of group 2 are able to either neutralize or compensate for the effects of TLF.[27]
In contrast T. b. rhodesiense is dependent upon the expression of a serum resistance associated (SRA) gene.[28] This gene is not found in T. b. gambiense.[29]
Genome
The genome of T. brucei is made up of:[30]
- 11 pairs of large chromosomes of 1 to 6 megabase pairs.
- 3–5 intermediate chromosomes of 200 to 500 kilobase pairs.
- Around 100 minichromosomes of around 50 to 100 kilobase pairs. These may be present in multiple copies per haploid genome.
Most genes are held on the large chromosomes, with the minichromosomes carrying only VSG genes. The genome has been sequenced and is available online .
The mitochondrial genome is found condensed into the kinetoplast, an unusual feature unique to the kinetoplastid protozoans. The kinetoplast and the basal body of the flagellum are strongly associated via a cytoskeletal structure.
In 1993, a new base, beta-d-glucopyranosyloxymethyluracil (base J), was identified in the nuclear DNA of T. brucei.[31]
VSG coat
The surface of the trypanosome is covered by a dense coat of ~5 x 106 molecules of variant surface glycoprotein (VSG).[32] This coat enables an infecting T. brucei population to persistently evade the host's immune system, allowing chronic infection. VSG is highly immunogenic, and an immune response raised against a specific VSG coat rapidly kills trypanosomes expressing this variant. Antibody-mediated trypanosome killing can also be observed in vitro by a complement-mediated lysis assay. However, with each cell division there is a possibility that one or both of the progeny will switch expression to change the VSG that is being expressed. The frequency of VSG switching has been measured to be approximately 0.1% per division.[33] As T. brucei populations can peak at a size of 1011 within a host [34] this rapid rate of switching ensures that the parasite population is typically highly diverse.[35][36] Because host immunity against a specific VSG does not develop immediately, some parasites will have switched to an antigenically-distinct VSG variant, and can go on to multiply and continue the infection. The clinical effect of this cycle is successive 'waves' of parasitemia (trypanosomes in the blood).[32]
Expression of VSG genes occurs through a number of mechanisms yet to be fully understood.[37] The expressed VSG can be switched either by activating a different expression site (and thus changing to express the VSG in that site), or by changing the VSG gene in the active site to a different variant. The genome contains many hundreds if not thousands of VSG genes, both on minichromosomes and in repeated sections ('arrays') in the interior of the chromosomes. These are transcriptionally silent, typically with omitted sections or premature stop codons, but are important in the evolution of new VSG genes. It is estimated up to 10% of the T. brucei genome may be made up of VSG genes or pseudogenes. It is thought that any of these genes can be moved into the active site by recombination for expression.[38]
Killing by human serum and resistance to human serum killing
Trypanosoma brucei brucei (as well as related species T. equiperdum and T. evansi) is not human infective because it is susceptible to innate immune system 'trypanolytic' factors present in the serum of some primates, including humans. These trypanolytic factors have been identified as two serum complexes designated trypanolytic factors (TLF-1 and -2) both of which contain haptoglobin related protein (HPR) and apolipoprotein LI (ApoL1). TLF-1 is a member of the high density lipoprotein family of particles while TLF-2 is a related high molecular weight serum protein binding complex.[39][40] The protein components of TLF-1 are haptoglobin related protein (HPR), apolipoprotein L-1 (apoL-1) and apolipoprotein A-1 (apoA-1). These three proteins are colocalized within spherical particles containing phospholipids and cholesterol. The protein components of TLF-2 include IgM and apolipoprotein A-I.
Trypanolytic factors are found only in a few species including humans, gorillas, mandrills, baboons and sooty mangabeys.[41] This appears to be because haptoglobin related protein and apolipoprotein L-1 are unique to primates.[41] This suggests these gene appeared in the primate genome 25 million years ago-35 million years ago.
Human infective subspecies T. b. gambiense and T. b. rhodesiense have evolved mechanisms of resisting the trypanolytic factors, described below.
ApoL1
ApoL1 is a member of a six gene family, ApoL1-6, that have arisen by tandem duplication. These proteins are normally involved in host apoptosis or autophagic death and possess a Bcl-2 homology domain 3.[42] ApoL1 has been identified as the toxic component involved in trypanolysis.[43] ApoLs have been subject to recent selective evolution possibly related to resistance to pathogens[44]
The gene encoding ApoL1 is found on the long arm of chromosome 22 (22q12.3). Variants of this gene, termed G1 and G2, provide protection against T. b. rhodesiense.[45] These benefits are not without their downside as a specific ApoL1 glomeropathy has been identified.[45][46] This glomeropathy may help to explain the greater prevalence of hypertension in African populations.[47]
The gene encodes a protein of 383 residues, including a typical signal peptide of 12 amino acids.[48] The plasma protein is a single chain polypeptide with an apparent molecular mass of 42 kiloDaltons. ApoL1 has a membrane pore forming domain functionally similar to that of bacterial colicins.[49] This domain is flanked by the membrane addressing domain and both these domains are required for parasite killing.
Within the kidney, ApoL1 is found in the podocytes in the glomeruli, the proximal tubular epithelium and the arteriolar endothelium.[50] It has a high affinity for phosphatidic acid and cardiolipin and can be induced by interferon gamma and tumor necrosis factor alpha.[51]
Hpr
Hpr is 91% identical to haptoglobin (Hp), an abundant acute phase serum protein, which possesses a high affinity for haemoglobin (Hb). When Hb is released from erythrocytes undergoing intravascular hemolysis Hp forms a complex with the Hb and these are removed from the circulation by the CD163 scavenger receptor. In contrast to Hp–Hb, the Hpr–Hb complex does not bind CD163 and the Hpr serum concentration appears to be unaffected by haemolysis.
Killing mechanism
The association HPR with haemoglobin allows TLF-1 binding and uptake via the trypanosome haptoglobin-hemoglobin receptor (TbHpHbR).[52] TLF-2 enters trypanosomes independently of TbHpHbR.[52] TLF-1 uptake is enhanced in the low levels of haptoglobin which competes with haptoglobin related protein to bind free haemoglobin in the serum. However the complete absence of haptoglobin is associated with a decreased killing rate by serum.[53]
The trypanosome haptoglobin-hemoglobin receptor is an elongated three a-helical bundle with a small membrane distal head.[54] This protein extends above the variant surface glycoprotein layer that surrounds the parasite.
The first step in the killing mechanism is the binding of TLF to high affinity receptors—the haptoglobin-hemoglobin receptors—that are located in the flagellar pocket of the parasite.[52][55] The bound TLF is endocytosed via coated vesicles and then trafficked to the parasite lysosomes. ApoL1 is the main lethal factor in the TLFs and kills trypanosomes after insertion into endosomal / lysosomal membranes.[43] After ingestion by the parasite, the TLF-1 particle is trafficked to the lysosome wherein Apo1 is activated by a pH mediated conformational change. After fusion with the lysosome the pH drops from ~7 to ~5. This induces a conformational change in the ApoL1 membrane addressing domain which in turn causes a salt bridge linked hinge to open. This releases ApoL1 from the HDL particle to insert in the lysosomal membrane. The ApoL1 protein then creates anionic pores in the membrane which leads to depolarization of the membrane, a continuous influx of chloride and subsequent osmotic swelling of the lysosome. This influx in its turn leads to rupture of the lysosome and the subsequent death of the parasite.[56]
Resistance mechanisms: T. b. gambiense
Trypanosoma brucei gambiense causes 97% of human cases of sleeping sickness. Resistance to ApoL1 is principally mediated by the hydrophobic ß-sheet of the T. b. gambiense specific glycoprotein.[57] Other factors involved in resistance appear to be a change in the cysteine protease activity and TbHpHbR inactivation due to a leucine to serine substitution (L210S) at codon 210.[57][58] This is due to a thymidine to cytosine mutation at the second codon position.
These mutations may have evolved due to the coexistence of malaria where this parasite is found.[57] Haptoglobin levels are low in malaria because of the haemolysis that occurs with the release of the merozoites into the blood. The rupture of the erythrocytes results in the release of free haem into the blood where it is bound by haptoglobin. The haem is then removed along with the bound haptoglobin from the blood by the reticuloendothelial system.
Resistance mechanisms: T. b. rhodesiense
Trypanosoma brucei rhodesiense relies on a different mechanism of resistance: the serum resistance associated protein (SRA). The SRA gene is a truncated version of the major and variable surface antigen of the parasite, the variant surface glycoprotein.[59] It has a low sequence homology with the VSGc (<25%). SRA is an expression site associated gene in T. b. rhodesiense and is located upstream of the VSGs in the active telomeric expression site.[60] The protein is largely localized to small cytoplasmic vesicles between the flagellar pocket and the nucleus. In T. b. rhodesiense the TLF is directed to SRA containing endosomes while some dispute remain on its presence in the lysosome.[43][61] SRA binds to ApoL1 using a coiled–coiled interaction at the ApoL1 SRA interacting domain while within the trypanosome lysosome.[43] This interaction prevents the release of the ApoL1 protein and the subsequent lysis of the lysosome and death of the parasite.
Baboons are known to be resistant to Trypanosoma brucei rhodesiense. The baboon version of the ApoL1 gene differs from the human gene in a number of respects including two critical lysines near the C terminus that are necessary and sufficient to prevent baboon ApoL1 binding to SRA.[62] Experimental mutations allowing ApoL1 to be protected from neutralization by SRA have been shown capable of conferring trypanolytic activity on T. b. rhodesiense.[28] These mutations resemble those found in baboons, but also resemble natural mutations conferring protection of humans against T. b. rhodesiense which are linked to kidney disease .[45]
See also
- List of parasites (human)
- Tryptophol, a chemical compound produced by the T. brucei which induces sleep in humans[63]
- David Bruce (1855–1931), a Scottish pathologist and microbiologist who investigated the Malta-fever and trypanosomes, identifying the cause of sleeping sickness.
- Simon Gaskell, professor of chemistry and current principal of Queen Mary, University of London, researches various forms of mass spectrometry to determine the quantity and longevity of these proteins.
References
- ↑ Baker, J.R. (1995). "The subspecific taxonomy of Trypanosoma brucei". Parasite. 2 (1): 3–12. ISSN 1252-607X. PMID 9137639. doi:10.1051/parasite/1995021003.

- ↑ Deborggraeve, Stijn; Koffi, Mathurin; Jamonneau, Vincent; Bonsu, Frank A.; Queyson, Richard; Simarro, Pere P.; Herdewijn, Piet; Büscher, Philippe (August 2008). "Molecular analysis of archived blood slides reveals an atypical human Trypanosoma infection". Diagnostic Microbiology and Infectious Disease. 61 (4): 428–433. PMID 18455900. doi:10.1016/j.diagmicrobio.2008.03.006.
- ↑ Masocha W, Kristensson K (2012). "Passage of parasites across the blood-brain barrier". Virulence. 3 (2): 202–12. PMC 3396699
 . PMID 22460639. doi:10.4161/viru.19178.
. PMID 22460639. doi:10.4161/viru.19178. - ↑ Legros, D; Ollivier, G; Gastellu-Etchegorry, M; Paquet, C; Burri, C; Jannin, J; Büscher, P (2002). "Treatment of human African trypanosomiasis--present situation and needs for research and development". The Lancet Infectious Diseases. 2 (7): 437–40. PMID 12127356. doi:10.1016/S1473-3099(02)00321-3.
- ↑ Gibson, W. C. (2007). "Resolution of the species problem in African trypanosomes". Int J Parasitol. 37 (8–9): 829–838. PMID 17451719. doi:10.1016/j.ijpara.2007.03.002.
- ↑ Joubert, JJ; Schutte, CH; Irons, DJ; Fripp, PJ (1993). "Ubombo and the site of David Bruce's discovery of Trypanosoma brucei". Transactions of the Royal Society of Tropical Medicine and Hygiene. 87 (4): 494–5. PMID 8249096. doi:10.1016/0035-9203(93)90056-v.
- ↑ Cook, GC (1994). "Sir David Bruce's elucidation of the aetiology of nagana--exactly one hundred years ago". Transactions of the Royal Society of Tropical Medicine and Hygiene. 88 (3): 257–8. PMID 7974656. doi:10.1016/0035-9203(94)90068-x.
- 1 2 Barrett MP, Burchmore RJ, Stich A, et al. (November 2003). "The trypanosomiases". Lancet. 362 (9394): 1469–80. PMID 14602444. doi:10.1016/S0140-6736(03)14694-6.
- ↑ Stephens NA, Kieft R, Macleod A, Hajduk SL (December 2012). "Trypanosome resistance to human innate immunity: targeting Achilles' heel". Trends Parasitol. 28 (12): 539–45. PMID 23059119. doi:10.1016/j.pt.2012.09.002.
- 1 2 3 4 "African animal trypanosomes". Food and Agricultural Organization. Retrieved 28 January 2016.
- 1 2 "African Trypanosomiasis". Davidson College. Retrieved 28 January 2016.
- 1 2 Langousis, Gerasimos; Hill, Kent L. (2014). "Motility and more: the flagellum of Trypanosoma brucei". Nature Reviews Microbiology. 12 (7): 505–518. PMC 4278896 . PMID 24931043. doi:10.1038/nrmicro3274.
- 1 2 3 4 Chatterjee, K.D. (2009). Parasitology (Protozoology and Helminthology) in relation to clinical medicine (13 ed.). New Delhi: CBC Publishers. pp. 56–57. ISBN 978-8-12-39-1810-5.
- 1 2 "Parasites - African Trypanosomiasis (also known as Sleeping Sickness)". Centers for Disease Control and Prevention. Retrieved 29 January 2016.
- ↑ Zampetti-Bosseler, F; Schweizer, J; Pays, E; Jenni, L; Steinert, M (1986). "Evidence for haploidy in metacyclic forms of Trypanosoma brucei". Proceedings of the National Academy of Sciences of the United States of America. 83 (16): 6063–4. PMC 386438 . PMID 3461475. doi:10.1073/pnas.83.16.6063.
- ↑ Jenni, L (1990). "Sexual stages in trypanosomes and implications". Annales de Parasitologie Humaine et Comparée. 65 Suppl 1: 19–21. PMID 2264676.
- ↑ Peacock, L.; Ferris, V.; Sharma, R.; Sunter, J.; Bailey, M.; Carrington, M.; Gibson, W. (2011). "Identification of the meiotic life cycle stage of Trypanosoma brucei in the tsetse fly". Proceedings of the National Academy of Sciences. 108 (9): 3671–3676. PMC 3048101 . PMID 21321215. doi:10.1073/pnas.1019423108.
- ↑ Peacock, Lori; Bailey, Mick; Carrington, Mark; Gibson, Wendy (January 2014). "Meiosis and Haploid Gametes in the Pathogen Trypanosoma brucei". Current Biology. 24 (2): 181–186. PMC 3928991 . PMID 24388851. doi:10.1016/j.cub.2013.11.044.
- ↑ Peacock, Lori; Ferris, Vanessa; Bailey, Mick; Gibson, Wendy (2014). "Mating compatibility in the parasitic protist Trypanosoma brucei". Parasites & Vectors. 7 (1): 78. PMC 3936861 . PMID 24559099. doi:10.1186/1756-3305-7-78.
- ↑ Hampl V, Hug L, Leigh JW, Dacks JB, Lang BF, Simpson AG, Roger AJ (March 2009). "Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic "supergroups"". Proc. Natl. Acad. Sci. U.S.A. 106 (10): 3859–64. PMC 2656170 . PMID 19237557. doi:10.1073/pnas.0807880106.
- ↑ Malik SB, Pightling AW, Stefaniak LM, Schurko AM, Logsdon JM (2008). "An expanded inventory of conserved meiotic genes provides evidence for sex in Trichomonas vaginalis". PLoS ONE. 3 (8): e2879. PMC 2488364 . PMID 18663385. doi:10.1371/journal.pone.0002879.
- ↑ Krisnky, Wiiliam L. (2009). "Tsetse fly (Glossinidae)". In Mullen, Gary R.; Durden, Lance. Medical and Veterinary Entomology (2 ed.). Amsterdam: Elsevier. p. 296. ISBN 978-0-0-80-91969-0.
- ↑ "African Trypanosomes: epidemiology and risk factors". Centers for Disease Control.
- ↑ Rocha, G; Martins, A; Gama, G; Brandão, F; Atouguia, J (2004). "Possible cases of sexual and congenital transmission of sleeping sickness". The Lancet. 363 (9404): 247. PMID 14738812. doi:10.1016/S0140-6736(03)15345-7.
- ↑ Weir W, Capewell P, Foth B, Clucas C, Pountain A, Steketee P, Veitch N, Koffi M, De Meeûs T, Kaboré J, Camara M, Cooper A, Tait A, Jamonneau V, Bucheton B, Berriman M, MacLeod A (2016) Population genomics reveals the origin and asexual evolution of human infective trypanosomes. Elife 5. pii: e11473. doi:10.7554/eLife.11473
- ↑ Paindavoine P, Pays E, Laurent M, et al. (February 1986). "The use of DNA hybridization and numerical taxonomy in determining relationships between Trypanosoma brucei stocks and subspecies". Parasitology. 92 (Pt 1): 31–50. PMID 3960593. doi:10.1017/S0031182000063435.
- ↑ Capewell P, Veitch NJ, Turner CM, et al. (September 2011). "Differences between Trypanosoma brucei gambiense groups 1 and 2 in their resistance to killing by trypanolytic factor 1". PLoS Negl Trop Dis. 5 (9): e1287. PMC 3167774 . PMID 21909441. doi:10.1371/journal.pntd.0001287.
- 1 2 Lecordier L, Vanhollebeke B, Poelvoorde P, Tebabi P, Andris F, Lins L and Pays E (2009). Mansfield, John M, ed. "C-terminal mutants of apolipoprotein L-I efficiently kill both Trypanosoma brucei brucei and Trypanosoma brucei rhodesiense". PLoS Pathog. 5 (12): e1000685. PMC 2778949 . PMID 19997494. doi:10.1371/journal.ppat.1000685.
- ↑ De Greef C, Imberechts H, Matthyssens G, Van Meirvenne N, Hamers R (September 1989). "A gene expressed only in serum-resistant variants of Trypanosoma brucei rhodesiense". Mol. Biochem. Parasitol. 36 (2): 169–76. PMID 2528066. doi:10.1016/0166-6851(89)90189-8.
- ↑ Ogbadoyi E, Ersfeld K, Robinson D, Sherwin T, Gull K (March 2000). "Architecture of the Trypanosoma brucei nucleus during interphase and mitosis". Chromosoma. 108 (8): 501–13. PMID 10794572. doi:10.1007/s004120050402.
- ↑ Borst P, Sabatini R (2008). "Base J: discovery, biosynthesis, and possible functions". Annu Rev Microbiol. 62: 235–51. PMID 18729733. doi:10.1146/annurev.micro.62.081307.162750.
- 1 2 Barry JD, McCulloch R (2001). "Antigenic variation in trypanosomes: enhanced phenotypic variation in a eukaryotic parasite". Adv Parasitol. 49: 1–70. ISBN 978-0-12-031749-3. PMID 11461029. doi:10.1016/S0065-308X(01)49037-3.
- ↑ Turner CM (August 1997). "The rate of antigenic variation in fly-transmitted and syringe-passaged infections of Trypanosoma brucei". FEMS Microbiol Lett. 153 (1): 227–31. PMID 9252591. doi:10.1111/j.1574-6968.1997.tb10486.x.
- ↑ Barry, J. D.; Hall, Plenderleith (2012). "Genome hyperevolution and the success of a parasite". Ann N Y Acad Sci. 1267 (1): 11–17. PMC 3467770 . PMID 22954210. doi:10.1111/j.1749-6632.2012.06654.x.
- ↑ Hall, James P. J.; Wang, Huanhuan; Barry, J. David; Horn, David (11 July 2013). "Mosaic VSGs and the Scale of Trypanosoma brucei Antigenic Variation". PLoS Pathogens. 9 (7): e1003502. PMC 3708902 . PMID 23853603. doi:10.1371/journal.ppat.1003502.
- ↑ Mugnier, M. R.; Cross, G. A. M.; Papavasiliou, F. N. (26 March 2015). "The in vivo dynamics of antigenic variation in Trypanosoma brucei". Science. 347 (6229): 1470–1473. PMC 4514441 . PMID 25814582. doi:10.1126/science.aaa4502.
- ↑ Pays E (November 2005). "Regulation of antigen gene expression in Trypanosoma brucei". Trends Parasitol. 21 (11): 517–20. PMID 16126458. doi:10.1016/j.pt.2005.08.016.
- ↑ Morrison, Liam J.; Marcello, Lucio; McCulloch, Richard (1 December 2009). "Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity". Cellular Microbiology. 11 (12): 1724–1734. PMID 19751359. doi:10.1111/j.1462-5822.2009.01383.x.
- ↑ Hajduk SL, Moore DR, Vasudevacharya J, et al. (March 1989). "Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein". J. Biol. Chem. 264 (9): 5210–7. PMID 2494183.
- ↑ Raper J, Fung R, Ghiso J, Nussenzweig V, Tomlinson S (April 1999). "Characterization of a novel trypanosome lytic factor from human serum". Infect. Immun. 67 (4): 1910–6. PMC 96545 . PMID 10085035.
- 1 2 Lugli EB, Pouliot M, Portela Mdel P, Loomis MR, Raper J (November 2004). "Characterization of primate trypanosome lytic factors". Mol. Biochem. Parasitol. 138 (1): 9–20. PMID 15500911. doi:10.1016/j.molbiopara.2004.07.004.
- ↑ Vanhollebeke B, Pays E (2006). "The function of apolipoproteins L". Cell. Mol. Life Sci. 63 (17): 1937–1944. PMID 16847577. doi:10.1007/s00018-006-6091-x.
- 1 2 3 4 Vanhamme L, Paturiaux-Hanocq F, Poelvoorde P, Nolan DP, Lins L, Van den Abbeele J, Pays A, Tebabi P, Xong HV, Jacquet A, Moguilevsky N, Dieu M, Kane JP, De Baetselier P, Brasseur R, Pays E (2003). "Apolipoprotein L-I is the trypanosome lytic factor of human serum". Nature. 422 (6927): 83–87. Bibcode:2003Natur.422...83V. PMID 12621437. doi:10.1038/nature01461.
- ↑ Smith EE, Malik HS (May 2009). "The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions". Genome Res. 19 (5): 850–8. PMC 2675973 . PMID 19299565. doi:10.1101/gr.085647.108.
- 1 2 3 Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Knob AU, Bernhardy A, Hicks PJ, Appel GB, Nelson GW, Vanhollebeke B, Winkler CA, Kopp JB, Pays E, Pollak MR (2010). "Association of trypanolytic ApoL1 variants with kidney disease in African-Americans". Science. 329 (5993): 841–845. PMC 2980843 . PMID 20647424. doi:10.1126/science.1193032.
- ↑ Wasser WG, Tzur S, Wolday D, et al. (2012). "Population genetics of chronic kidney disease: the evolving story of APOL1". J. Nephrol. 25 (5): 603–18. PMID 22878977. doi:10.5301/jn.5000179.
- ↑ Lipkowitz MS, Freedman BI, Langefeld CD, et al. (January 2013). "Apolipoprotein L1 gene variants associate with hypertension-attributed nephropathy and the rate of kidney function decline in African Americans". Kidney Int. 83 (1): 114–20. PMC 3484228 . PMID 22832513. doi:10.1038/ki.2012.263.
- ↑ Duchateau PN, Pullinger CR, Orellana RE, et al. (October 1997). "Apolipoprotein L, a new human high density lipoprotein apolipoprotein expressed by the pancreas. Identification, cloning, characterization, and plasma distribution of apolipoprotein L". J. Biol. Chem. 272 (41): 25576–82. PMID 9325276. doi:10.1074/jbc.272.41.25576.
- ↑ Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, et al. (July 2005). "Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes". Science. 309 (5733): 469–72. PMID 16020735. doi:10.1126/science.1114566.
- ↑ Madhavan SM, O'Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR (November 2011). "APOL1 localization in normal kidney and nondiabetic kidney disease". J. Am. Soc. Nephrol. 22 (11): 2119–28. PMC 3231786 . PMID 21997392. doi:10.1681/ASN.2011010069.
- ↑ Zhaorigetu S, Wan G, Kaini R, Jiang Z, Hu CA (November 2008). "ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death". Autophagy. 4 (8): 1079–82. PMC 2659410 . PMID 18927493. doi:10.4161/auto.7066.
- 1 2 3 Vanhollebeke B, Demuylder G, Nielsen MJ, Pays A, Tebabi P, Dieu M, Raes M, Moestrup SK, Pays E (2008). "A haptoglobin-hemoglobin receptor conveys innate immunity to Trypanosoma brucei in humans". Science. 320 (5876): 677–681. PMID 18451305. doi:10.1126/science.1156296.
- ↑ Vanhollebeke B, Nielsen MJ, Watanabe Y, et al. (March 2007). "Distinct roles of haptoglobin-related protein and apolipoprotein L-I in trypanolysis by human serum". Proc. Natl. Acad. Sci. U.S.A. 104 (10): 4118–23. PMC 1820718 . PMID 17360487. doi:10.1073/pnas.0609902104.
- ↑ Higgins MK, Tkachenko O, Brown A, Reed J, Raper J, Carrington M (January 2013). "Structure of the trypanosome haptoglobin-hemoglobin receptor and implications for nutrient uptake and innate immunity". Proc. Natl. Acad. Sci. U.S.A. 110 (5): 1905–10. PMC 3562850 . PMID 23319650. doi:10.1073/pnas.1214943110.
- ↑ Green HP, Del Pilar Molina Portela M, St Jean EN, Lugli EB, Raper J (January 2003). "Evidence for a Trypanosoma brucei lipoprotein scavenger receptor". J. Biol. Chem. 278 (1): 422–7. PMID 12401813. doi:10.1074/jbc.M207215200.
- ↑ Pays E, Vanhollebeke B, Vanhamme L, Paturiaux-Hanocq F, Nolan DP, Pérez-Morga D (2006). "The trypanolytic factor of human serum". Nature Reviews Microbiology. 4 (6): 477–486. PMID 16710327. doi:10.1038/nrmicro1428.
- 1 2 3 Uzureau P, Uzureau S, Lecordier L, Fontaine F, Tebabi P, Homblé F, Grélard A, Zhendre V, Nolan D, Lins L, Crowet JM, Pays A, Felu C, Poelvoorde P, Vanhollebeke B, Moestrup SK, Lyngsø J, Pedersen JS, Mottram J, Dufourc EJ, Pérez-Morga D, Pays E (2013). "Mechanism of Trypanosoma gambiense resistance to human serum". Nature. 501 (7467): 430–4. PMID 23965626. doi:10.1038/nature12516.
- ↑ DeJesus E, Kieft R, Albright B, Stephens NA, Hajduk SL (2013). "A single amino acid substitution in the group 1 Trypanosoma brucei gambiense haptoglobin-hemoglobin receptor abolishes TLF-1 binding". PLoS Pathog. 9 (4): e1003317. PMC 3630162 . PMID 23637606. doi:10.1371/journal.ppat.1003317.
- ↑ Pays E, Vanhollebeke B (July 2008). "Mutual self-defence: the trypanolytic factor story". Microbes Infect. 10 (9): 985–9. PMID 18675374. doi:10.1016/j.micinf.2008.07.020.
- ↑ Xong HV, Vanhamme L, Chamekh M, et al. (December 1998). "A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense". Cell. 95 (6): 839–46. PMID 9865701. doi:10.1016/S0092-8674(00)81706-7.
- ↑ Shiflett AM, Faulkner SD, Cotlin LF, Widener J, Stephens N, Hajduk SL (2007). "African trypanosomes: intracellular trafficking of host defense molecules". J. Eukaryot. Microbiol. 54 (1): 18–21. PMID 17300512. doi:10.1111/j.1550-7408.2006.00228.x.
- ↑ Thomson R, Molina-Portela P, Mott H, Carrington M, Raper J (November 2009). "Hydrodynamic gene delivery of baboon trypanosome lytic factor eliminates both animal and human-infective African trypanosomes". Proc. Natl. Acad. Sci. U.S.A. 106 (46): 19509–14. PMC 2780755 . PMID 19858474. doi:10.1073/pnas.0905669106.
- ↑ Richard Seed, J.; Seed, T. M.; Sechelski, J. (1978). "The biological effects of tryptophol (indole-3-ethanol): Hemolytic, biochemical and behavior modifying activity". Comparative Biochemistry and Physiology C. 60 (2): 175–185. doi:10.1016/0306-4492(78)90091-6.
External links
![]() Media related to Trypanosoma brucei at Wikimedia Commons
Media related to Trypanosoma brucei at Wikimedia Commons
- "Trypanosomiasis, African (Trypanosoma brucei gambiense) (Trypanosoma brucei rhodesiense)". DPDx — Laboratory Identification of Parasitic Diseases of Public Health Concern. Centers for Disease Control and Prevention. 29 November 2013.
- "Trypanosoma brucei". NCBI Taxonomy Browser. 5691.