Super-resolution microscopy
Super-resolution microscopy is a form of light microscopy. Due to the diffraction of light, the resolution of conventional light microscopy is limited, as stated by Ernst Abbe in 1873.[1] A good approximation of the resolution attainable is the full width at half maximum (FWHM) of the point spread function, and a precise widefield microscope with high numerical aperture and visible light usually reaches a resolution of ~250 nm.
Super-resolution techniques allow images to be taken with a higher resolution than the diffraction limit.[2]
True subwavelength imaging techniques include those that utilize the Pendry Superlens and near field scanning optical microscopy, the 4Pi Microscope and structured illumination microscopy technologies like SIM and SMI. However, the majority of techniques of importance in biological imaging fall into the functional category.
There are two major groups of methods for functional super-resolution microscopy:[3]
- Deterministic super-resolution: The most commonly used emitters in biological microscopy, fluorophores, show a nonlinear response to excitation, and this nonlinear response can be exploited to enhance resolution. These methods include STED, GSD, RESOLFT and SSIM.
- Stochastic super-resolution: The chemical complexity of many molecular light sources gives them a complex temporal behavior, which can be used to make several close-by fluorophores emit light at separate times and thereby become resolvable in time. These methods include Super-resolution optical fluctuation imaging (SOFI) and all single-molecule localization methods (SMLM) such as SPDM, SPDMphymod, PALM, FPALM, STORM and dSTORM.
On October 8, 2014, the Nobel Prize in Chemistry was awarded to Eric Betzig, W.E. Moerner and Stefan Hell for "the development of super-resolved fluorescence microscopy," which brings "optical microscopy into the nanodimension".[4][5]
History
In 1978, the first theoretical ideas had been developed to break the Abbe limit using a 4Pi microscope as a confocal laser scanning fluorescence microscope where the light is focused ideally from all sides to a common focus that is used to scan the object by 'point-by-point' excitation combined with 'point-by-point' detection.[6]
Some of the following information was gathered (with permission) from a chemistry blog's review of sub-diffraction microscopy techniques Part I and Part II. For a review, see also reference.[7]
In 1986, the super-resolution optical microscope based on stimulated emission was patented by Okhonin.[8]
True super-resolution techniques
Near-field scanning optical microscope (NSOM)
Local enhancement / ANSOM / optical nano-antennas
Near-field optical random mapping (NORM) microscopy
NORM (near-field optical random mapping) microscopy is a method of optical near-field acquisition by a far-field microscope through the observation of nanoparticles' Brownian motion in an immersion liquid.[14][15]
NORM utilizes object surface scanning by stochastically moving nanoparticles. Through the microscope, nanoparticles look like symmetric round spots. The spot width is equivalent to the point spread function (~ 250 nm) and is defined by the microscope resolution. Lateral coordinates of the given particle can be evaluated with a precision much higher than the resolution of the microscope. By collecting the information from many frames one can map out the near field intensity distribution across the whole field of view of the microscope. In comparison with NSOM and ANSOM this method does not require any special equipment for tip positioning and has a large field of view and a depth of focus. Due to the large number of scanning "sensors" one can get shorter time of image acquisition.
4Pi
A 4Pi microscope is a laser scanning fluorescence microscope with an improved axial resolution. The typical value of 500–700 nm can be improved to 100–150 nm, which corresponds to an almost spherical focal spot with 5–7 times less volume than that of standard confocal microscopy.
The improvement in resolution is achieved by using two opposing objective lenses, both of which are focused to the same geometrical location. Also, the difference in optical path length through each of the two objective lenses is carefully aligned to be minimal. By this, molecules residing in the common focal area of both objectives can be illuminated coherently from both sides and also the reflected or emitted light can be collected coherently, i.e. coherent superposition of emitted light on the detector is possible. The solid angle that is used for illumination and detection is increased and approaches the ideal case. In this case the sample is illuminated and detected from all sides simultaneously.[16][17]

Up to now, the best quality in a 4Pi microscope was reached in conjunction with the STED principle in fixed cells [18] and RESOLFT microscopy with switchable proteins in living cells.[19]
Structured illumination microscopy (SIM)

There is also the wide-field structured-illumination (SI) approach.[20][21][22]
SI enhances spatial resolution by collecting information from frequency space outside the observable region. This process is done in reciprocal space: The Fourier transform (FT) of an SI image contains superimposed additional information from different areas of reciprocal space; with several frames with the illumination shifted by some phase, it is possible to computationally separate and reconstruct the FT image, which has much more resolution information. The reverse FT returns the reconstructed image to a super-resolution image.
A number of applications have recently surfaced, in which SIM microscopy could potentially replace electron microscopy as part of routine diagnosis. These include diagnosis of kidney disorders,[23] kidney cancer,[24] and blood diseases.[25]
- Images of cell nuclei and mitotic stages recorded with 3D-SIM.
 Comparison confocal microscopy – 3D-SIM
Comparison confocal microscopy – 3D-SIM Cell nucleus in prophase from various angles
Cell nucleus in prophase from various angles Two mouse cell nuclei in prophase.
Two mouse cell nuclei in prophase. mouse cell in telophase
mouse cell in telophase
Spatially modulated illumination (SMI)
One implementation of structured illumination is known as spatially modulated illumination. Like standard structured illumination, the SMI technique modifies the point spread function (PSF) of a microscope in a suitable manner. In this case however, "the optical resolution itself is not enhanced";[26] instead structured illumination is used to maximize the precision of distance measurements of fluorescent objects, to "enable size measurements at molecular dimensions of a few tens of nanometers".[26]
The Vertico SMI microscope achieves structured illumination by using one or two opposing interfering laser beams along the axis. The object being imaged is then moved in high-precision steps through the wave field, or the wave field itself is moved relative to the object by phase shift. This results in an improved axial size and distance resolution.[26][27][28]
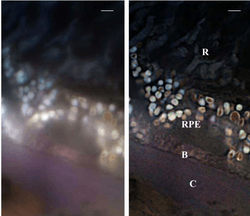
SMI can be combined with other super resolution technologies, for instance with 3D LIMON or LSI-TIRF as a total internal reflection interferometer with laterally structured illumination. This SMI technique allows one to acquire light-optical images of autofluorophore distributions in the sections from human eye tissue with previously unmatched optical resolution. Use of three different excitation wavelengths (488, 568 and 647 nm), enables one to gather spectral information about the autofluorescence signal. This has been used for human eye tissue affected by macular degeneration.[29]
Deterministic functional techniques
RESOLFT microscopy is an optical microscopy with very high resolution that can image details in samples that cannot be imaged with conventional or confocal microscopy. Within RESOLFT the principle of STED microscopy[30][31] and GSD microscopy are generalized. Also, there are techniques with other concepts than RESOLFT or SSIM, for example fluorescence microscopy using optical AND gate property of nitrogen-vacancy center.[32]
Stimulated emission depletion (STED)

STED (stimulated emission depletion microscopy) uses two laser pulses, the excitation pulse for excitation of the fluorophores to their fluorescent state and the STED pulse for the de-excitation of fluorophores by means of stimulated emission.[8][33][34][35][36][37] In practice, the excitation laser pulse is first applied whereby a STED pulse soon follows (But STED without pulses using continuous wave lasers is also used). Furthermore, the STED pulse is modified in such a way that it features a zero-intensity spot, which coincides with the excitation focal spot. Due to the non-linear dependence of the stimulated emission rate on the intensity of the STED beam, all the fluorophores around the focal excitation spot will be in their off state (the ground state of the fluorophores). By scanning this focal spot one retrieves the image. The FWHM of the PSF of the excitation focal spot can theoretically be compressed to an arbitrary width by raising the intensity of the STED pulse, according to equation (1).
- (1)

∆r is the lateral resolution, ∆ is the FWHM of the diffraction limited PSF, Imax is the peak intensity of the STED laser, and is the threshold intensity needed in order to achieve saturated emission depletion.

The main disadvantage of STED, which has prevented its widespread use, is that the machinery is complicated. On the one hand, the image acquisition speed is relatively slow for large fields of view because of the need to scan the sample in order to retrieve an image. On the other hand, it can be very fast for smaller fields of view: Recordings with up to 80 frames per second have been shown. [38][39] Due to a large Is value associated with STED, there is a need for a high-intensity excitation pulse, which may cause damage to the sample.
Ground state depletion (GSD)
GSD microscopy or ground state depletion microscopy, uses the triplet state of a fluorophore as the off-state and the singlet state as the on-state, whereby an excitation laser is used to drive the fluorophores at the periphery of the singlet state molecule to the triplet state. This is much like STED, where the off-state is the ground state of fluorophores, which is why equation (1) also applies in this case. The value is smaller than in STED making super-resolution imaging possible at a much smaller laser intensity. Compared to STED though, the fluorophores used in GSD are generally less photostable and the saturation of the triplet state may be harder to realize.[40]
Saturated structured illumination microscopy (SSIM)
SSIM (saturated structured-illumination microscopy) exploits the nonlinear dependence of the emission rate of fluorophores on the intensity of the excitation laser.[41] By applying a sinusoidal illumination pattern with a peak intensity close to that needed in order to saturate the fluorophores in their fluorescent state one retrieves Moiré fringes. The fringes contain high order spatial information that may be extracted by computational techniques. Once the information is extracted a super-resolution image is retrieved.
SSIM requires shifting the illumination pattern multiple times, effectively limiting the temporal resolution of the technique. In addition there is a need of very photostable fluorophores due to the saturating conditions. These conditions also induce radiation damage to the sample that restricts the possible applications in which SSIM may be used.
Examples for this microscopy are shown under section Structured illumination microscopy (SIM), Images of cell nuclei and mitotic stages recorded with 3D-SIM Microscopy.
Stochastic functional techniques
Localization microscopy
Single-molecule localization microscopy (SMLM) summarizes all microscopical techniques that achieve super-resolution by isolating emitters and fitting their images with the point spread function (PSF). Normally, the width of the point spread function (~ 250 nm) limits resolution. However, given an isolated emitter, one is able to determine its location with a precision only limited by its intensity according to equation (2).
- (2)

Here, Δloc is the localization precision, Δ is the FWHM of the PSF and N is the number of collected photons. This fitting process can only be performed reliably for isolated emitters (see Deconvolution), and interesting biological samples are so densely labeled with emitters that fitting is impossible when all emitters are active at the same time. SMLM techniques solve this dilemma by activating only a sparse subset of emitters at the same time, localizing these few emitters very precisely, deactivating them and activating another subset.
Generally, localization microscopy is performed with fluorophores. Suitable fluorophores reside in a non-fluorescent dark state for most of the time and are activated stochastically, typically with an excitation laser of low intensity. A readout laser stimulates fluorescence and bleaches or photoswitches the fluorophores back to a dark state, typically within 10–100 ms. The photons emitted during the fluorescent phase are collected with a camera and the resulting image of the fluorophore (which is distorted by the PSF) can be fitted with very high precision even on the order of a few Angstroms.[42] Repeating the process several thousand times ensures that all fluorophores can go through the bright state and are recorded. A computer then reconstructs a super-resolved image.
The desirable traits of fluorophores used for these methods, in order to maximize the resolution, are that they should be bright. That is, they should have a high extinction coefficient and a high quantum yield. They should also possess a high contrast ratio (ratio between the number of photons emitted in the light state and the number of photons emitted in the dark state). Also, a densely labeled sample is desirable according to the Nyquist criteria.
The multitude of localization microscopy methods differ mostly in the type of fluorophores used.
Localization microscopy SPDM

A single, tiny source of light can be located much better than the resolution of a microscope: Although the light will produce a blurry spot, computer algorithms can be used to accurately calculate the center of the blurry spot, taking into account the point spread function of the microscope, the noise properties of the detector, and so on. However, this approach does not work when there are too many sources close to each other: The sources all blur together.
SPDM (spectral precision distance microscopy) is a family of techniques in fluorescence microscopy which gets around this problem by measuring just a few sources at a time, so that each source is "optically isolated" from the others (i.e., separated by more than the microscope's resolution, typically ~200-250 nm).[43][44][45] Then, the above technique (finding the center of each blurry spot) can be used.
If the molecules have a variety of different spectra (absorption spectra and/or emission spectra), then it is possible to look at light from just a few molecules at a time by using the appropriate light sources and filters. Molecules can also be distinguished in more subtle ways based on fluorescent lifetime and other techniques.[43]
The structural resolution achievable using SPDM can be expressed in terms of the smallest measurable distance between two in their spatial position determined punctiform particle of different spectral characteristics ("topological resolution"). Modeling has shown that under suitable conditions regarding the precision of localization, particle density etc., the "topological resolution" corresponds to a "space frequency" which in terms of the classical definition is equivalent to a much improved optical resolution.
SPDM is a localization microscopy which achieves an effective optical resolution several times better than the conventional optical resolution (approx. 200-250 nm), represented by the half-width of the main maximum of the effective point image function. By applying suitable laser optical precision processes, position and distances significantly smaller than the half-width of the point spread function (conventionally 200-250 nm) can be measured with nanometer accuracy between targets with different spectral signatures.[43] An important area of application is genome research (study of the functional organization of the genome). Another important area of use is research into the structure of membranes.
SPDMphymod
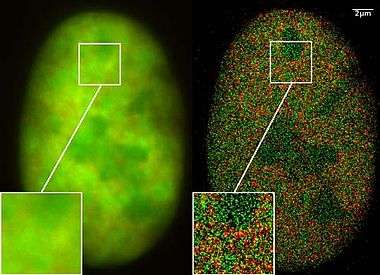
Localization microscopy for many standard fluorescent dyes like GFP, Alexa dyes and fluorescein molecules is possible if certain photo-physical conditions are present. With this so-called SPDMphymod (physically modifiable fluorophores) technology a single laser wavelength of suitable intensity is sufficient for nanoimaging[46] in contrast to other localization microscopy technologies that need two laser wavelengths when special photo-switchable/photo-activatable fluorescence molecules are used. A further example for the use of SPDMphymod is an analysis of Tobacco mosaic virus (TMV) particles[47] or the study of virus–cell interaction.[48][49]
Based on singlet triplet state transitions it is crucial for SPDMphymod that this process is ongoing and leading to the effect that a single molecule comes first into a very long-living reversible dark state (with half-life of several seconds even) from which it returns to a fluorescent state emitting many photons for several milliseconds before it returns into a very long-living so-called irreversible dark state. SPDMphymod microscopy uses fluorescent molecules that are emitting the same spectral light frequency but with different spectral signatures based on the flashing characteristics. By combining two thousands images of the same cell, it is possible using laser optical precision measurements to record localization images with significantly improved optical resolution.[50]
Standard fluorescent dyes already successfully used with the SPDMphymod technology are GFP, RFP, YFP, Alexa 488, Alexa 568, Alexa 647, Cy2, Cy3, Atto 488 and fluorescein.
Cryogenic Optical Localization in 3D (COLD)

Cryogenic Optical Localization in 3D (COLD) is a method that allows localizing multiple fluorescent sites within a single small- to medium-sized biomolecule with Angstrom-scale resolution.[42] The localization precision in this approach is enhanced because the slower photochemistry at low temperatures leads to higher number of photons that can be emitted from each fluorophore before photobleaching.[51][52] As a result, cryogenic stochastic localization microscopy reaches the required sub-molecular resolution to resolve the 3D positions of several fluorophores attached to a small protein. By employing algorithms known from electron microscopy, the 2D projections of fluorophores are reconstructed into a 3D configuration. COLD brings fluorescence microscopy to its fundamental limit given by the size of the label. The method can also be combined with other structural biology techniques such as X-ray crystallography, magnetic resonance spectroscopy and electron microscopy to provide valuable complementary information and specificity.
fBALM

fBALM (Activated Localization Microscopy based on DNA structure fluctuations) is a general concept for Single Molecule localization Microscopy (SMLM) super-resolved imaging of DNA-binding dyes based on modifying the properties of DNA and the dye. By careful adjustment of the chemical environment leading to local, reversible DNA melting and hybridization control over the fluorescence signal of the DNA-binding dye molecules can be introduced. A transient binding as the basis for the variation of binding-activated localization microscopy (BALM) has been proposed. Intercalating and minor-groove binding DNA dyes can be used to register (optically isolate) only a few DNA-binding dye signals at a time. This DNA structure fluctuation-assisted BALM (fBALM) has been used to nanoscale differences in nuclear architecture with an anticipated structural resolution of approximately 50 nm. Imaging chromatin nanostructure with binding-activated localization microscopy based on DNA structure fluctuations.[53]
STORM, PALM and FPALM
Stochastic optical reconstruction microscopy (STORM), photo activated localization microscopy (PALM) and fluorescence photo-activation localization microscopy (FPALM) are super-resolution imaging techniques that utilize sequential activation and time-resolved localization of photoswitchable fluorophores to create high resolution images. During imaging, only an optically resolvable subset of fluorophores is activated to a fluorescent state at any given moment, such that the position of each fluorophore can be determined with high precision by finding the centroid position of the single-molecule images of particular fluorophore. The fluorophore is subsequently deactivated, and another subset is activated and imaged. Iteration of this process allows numerous fluorophores to be localized and a super-resolution image to be constructed from the image data. These three methods were published independently during a short period of time and their principle is identical. STORM was originally described using Cy5 and Cy3 dyes attached to nucleic acids or proteins,[54] while PALM and FPALM was described using photoswitchable fluorescent proteins.[55][56] In principle any photoswitchable fluorophore can be used, and STORM has been demonstrated with a variety of different probes and labeling strategies. Using stochastic photoswitching of single fluorophores, such as Cy5,[57] STORM can be performed with a single red laser excitation source. The red laser both switches the Cy5 fluorophore to a dark state by formation of an adduct[58][59] and subsequently returns the molecule to the fluorescent state. Many other dyes have been also used for STORM.[60][61][62][63][64][65] In addition to single fluorophores, dye-pairs consisting of an activator fluorophore (such as Alexa 405, Cy2, and Cy3) and a photoswitchable reporter dye (such as Cy5, Alexa 647, Cy5.5, and Cy7) can be used for STORM.[54][66][67] In this scheme, the activator fluorophore, when excited near its absorption maximum, serves to reactivate the photoswitchable dye to the fluorescent state. Multicolor imaging has been performed by using different activation wavelengths to distinguish dye-pairs based on the activator fluorophore used[66][67][68] or using spectrally distinct photoswitchable fluorophores either with or without activator fluorophores.[60][69][70] Photoswitchable fluorescent proteins can be used as well.[55][56][70][71] Highly specific labeling of biological structures with photoswitchable probes has been achieved with antibody staining,[66][67][68][72] direct conjugation of proteins,[73] and genetic encoding.[55][56][70][71] STORM has also been extended to three-dimensional imaging using optical astigmatism, in which the elliptical shape of the point spread function encodes the x, y, and z positions for samples up to several micrometers thick,[67][72] and has been demonstrated in living cells.[70] To date, the spatial resolution achieved by this technique is ~20 nm in the lateral dimensions and ~50 nm in the axial dimension and the temporal resolution is as fast as 0.1–0.33s.
Label-free localization microscopy

Optical resolution of cellular structures in the range of about 50 nm can be achieved even in label-free cells using the localization microscopy SPDM.
Beside a substantial resolution improvement of autofluorescent structures, using two different laser wavelengths SPDM reveals cellular objects which are not detectable under conventional fluorescence wide-field imaging conditions.
As a control the positions of the detected objects in the localization image match these in the bright-field image.[74]
Label-free superresolution microscopy has also been demonstrated using the fluctuations of surface enhanced Raman scattering signal on a highly uniform plasmonic metasurface. [75]
Direct stochastical optical reconstruction microscopy (dSTORM)
dSTORM utilizes the photoswitching of a single fluorophore. In dSTORM, fluorophores are embedded in an oxidizing and reducing buffer system (ROXS) and fluorescence is excited. Sometimes, stochastically, the fluorophore will enter a triplet or some other dark state which is sensitive to the oxidation state of the buffer. As the molecules return stochastically they can be excited to fluoresce so that single molecule positions can be recorded.[76] Development of the dSTORM method occurred in 3 independent laboratories at about the same time and was called 'Reversible photobleaching microscopy -RPM",[77] "Ground state depletion microscopy followed by individual molecule return" -GSDIM[78] as well as the now generally accepted moniker dSTORM.[79]
Software for localization microscopy [80]
Localization microscopy depends heavily on software that can precisely fit the point spread function (PSF) to millions of images of active fluorophores within a few minutes. Since the classical analysis methods and software suites used in natural sciences are too slow to computationally solve these problems, often taking hours of computation for processing data measured in minutes, specialised software programs have been developed. Many of these localisation software packages are open-source, the are listed at SMLM Software Benchmark. Once molecule positions have been determined, the locations need to be displayed and several algorithms for display have been developed e.g.[81]
Super-resolution optical fluctuation imaging (SOFI)
It is possible to circumvent the need for PSF fitting inherent in single molecule localization microscopy (SMLM) by directly computing the temporal autocorrelation of pixels. This technique is called super-resolution optical fluctuation imaging (SOFI) and has been shown to be more precise than SMLM when the density of concurrently active fluorophores is very high.
Combination of techniques
3D light microscopical nanosizing (LIMON) microscopy

3 D LIMON (Light MicrOscopical nanosizing microscopy) images using the Vertico SMI microscope are made possible by the combination of SMI and SPDM, whereby first the SMI and then the SPDM process is applied.
The SMI process determines the center of particles and their spread in the direction of the microscope axis. While the center of particles/molecules can be determined with a 1–2 nm precision, the spread around this point can be determined down to an axial diameter of approx. 30–40 nm.
Subsequently, the lateral position of the individual particle/molecule is determined using SPDM, achieving a precision of a few nanometers.[82]
As a biological application in the 3D dual color mode the spatial arrangements of Her2/neu and Her3 clusters was achieved. The positions in all three directions of the protein clusters could be determined with an accuracy of about 25 nm.[83]
Integrated correlative light and electron microscopy
Combining a super-resolution microscope with an electron microscope enables the visualization of contextual information with the labelling provided by fluorescence markers. This overcomes the problem of the black backdrop that the researcher is left with when using only a light microscope. In an integrated system, the sample is measured by both microscopes simultaneously.[84]
See also
- Multifocal plane microscopy (MUM)
- Stimulated emission depletion microscope (STED)
- Photoactivated localization microscopy (PALM)
- Stochastic optical reconstruction microscopy (STORM)
- Deconvolution
- Photoactivatable probes
- Correlative Light-Electron Microscopy
References
- ↑ Abbe, E. (1873). "Beitrage zur Theorie des Mikroskops und der mikroskopischen Wahrmehmung". Archiv für mikroskopische Anatomie (in German). 9: 413–420. doi:10.1007/BF02956173.
- ↑ Neice, A. (2010). "Methods and Limitations of Subwavelength Imaging". Advances in Imaging and Electron Physics. Advances in Imaging and Electron Physics. 163: 117–140. ISBN 978-0-12-381314-5. doi:10.1016/S1076-5670(10)63003-0.
- ↑ SPIE (March 2015). "W.E. Moerner plenary presentation: Single-molecule spectroscopy, imaging, and photocontrol -- foundations for super-resolution microscopy". SPIE Newsroom. doi:10.1117/2.3201503.17.
- ↑ Ritter, Karl; Rising, Malin (8 October 2014). "2 Americans, 1 German win chemistry Nobel". Associated Press. Retrieved 8 October 2014.
- ↑ Chang, Kenneth (8 October 2014). "2 Americans and a German Are Awarded Nobel Prize in Chemistry". New York Times. Retrieved 8 October 2014.
- ↑ Cremer C, Cremer T (September 1978). "Considerations on a laser-scanning-microscope with high resolution and depth of field". Microsc Acta. 81 (1): 31–44. PMID 713859.
- ↑ WEM News and Views
- 1 2 V.A. Okhonin, Method of investigating specimen microstructure, Patent SU 1374922, priority date April 10, 1986, Published on July 30, 1991, Soviet Patents Abstracts, Section EI, Week 9218, Derwent Publications Ltd., London, GB; Class S03, p. 4. Cited by patents US 5394268 A (1993) and US RE38307 E1 (1995). From the English translation: "The essence of the invention is as follows. Luminescence is excited in a sample placed in the field of several standing light waves, which cause luminescence quenching because of stimulated transitions...".
- ↑ "Fresnel Diffraction Applet" (Java applet). Retrieved 2007-08-22.
- ↑ Cummings JR, Fellers TJ, Davidson MW (2007). "Specialized Microscopy Techniques – Near-Field Scanning Optical Microscopy". Olympus Microscopy Resource Center. Archived from the original on 14 March 2007. Retrieved 2007-08-22.
- ↑ Sánchez EJ, Novotny L, Xie XS (1999). "Near-Field Fluorescence Microscopy Based on Two-Photon Excitation with Metal Tips". Phys Rev Lett. 82 (20): 4014–7. Bibcode:1999PhRvL..82.4014S. doi:10.1103/PhysRevLett.82.4014.
- ↑ Schuck PJ, Fromm DP, Sundaramurthy A, Kino GS, Moerner WE (2005). "Improving the Mismatch between Light and Nanoscale Objects with Gold Bowtie Nanoantennas". Phys Rev Lett. 94 (1): 017402. Bibcode:2005PhRvL..94a7402S. PMID 15698131. doi:10.1103/PhysRevLett.94.017402.
- ↑ Muhlschlegel P, Eisler HJ, Martin OJ, Hecht B, Pohl DW (2005). "Resonant optical antennas". Science. 308 (5728): 1607–9. Bibcode:2005Sci...308.1607M. PMID 15947182. doi:10.1126/science.1111886.
- ↑ US patent 2009/0116,024, priority date 7 April 2006: J. V. Mikliaev, S. A. Asselborn Method for obtaining a high resolution image
- ↑ Yu. V. Miklyaev; S. A. Asselborn; K. A. Zaytsev; M. Ya. Darscht (2014). "Superresolution microscopy in far-field by near-field optical random mapping nanoscopy". Appl. Phys. Lett. 105 (11): 113103(1–4). Bibcode:2014ApPhL.105k3103M. doi:10.1063/1.4895922.
- ↑ Cremer, Christoph; Cremer, Thomas (1978). "Considerations on a laser-scanning-microscope with high resolution and depth of field" (PDF). M1Croscopica Acta. 81 (1): 31–44.
- ↑ S.W. Hell; E.H.K. Stelzer; S. Lindek; C. Cremer (1994). "Confocal microscopy with an increased detection aperture: type-B 4Pi confocal microscopy". Optics Letters. 19 (3): 222–224. Bibcode:1994OptL...19..222H. PMID 19829598. doi:10.1364/OL.19.000222.
- ↑ R. Schmidt; C.A. Wurm; S. Jakobs; J. Engelhardt; A. Egner; S.W. Hell (2008). "Spherical nanosized focal spot unravels the interior of cells" (PDF). Nature Methods. 5 (6): 539–544. PMID 18488034. doi:10.1038/nmeth.1214.
- ↑ Ulrike Böhm; Stefan W. Hell; Roman Schmidt (2016). "4Pi-RESOLFT nanoscopy". Nature Communications. 7 (10504): 1–8. Bibcode:2016NatCo...710504B. PMC 4740410
 . PMID 26833381. doi:10.1038/ncomms10504.
. PMID 26833381. doi:10.1038/ncomms10504. - ↑ Bailey B, Farkas DL, Taylor DL, Lanni F (November 1993). "Enhancement of axial resolution in fluorescence microscopy by standing-wave excitation". Nature. 366 (6450): 44–8. Bibcode:1993Natur.366...44B. PMID 8232536. doi:10.1038/366044a0.
- ↑ Gustafsson MG (May 2000). "Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy". J Microsc. 198 (Pt 2): 82–7. PMID 10810003. doi:10.1046/j.1365-2818.2000.00710.x.
- ↑ Gustafsson MG (September 2005). "Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution". Proc. Natl. Acad. Sci. U.S.A. 102 (37): 13081–6. Bibcode:2005PNAS..10213081G. PMC 1201569 . PMID 16141335. doi:10.1073/pnas.0406877102.
- ↑ Pullman, James M.; Nylk, Jonathan; Campbell, Elaine C.; Gunn-Moore, Frank J.; Prystowsky, Michael B.; Dholakia, Kishan (2016-02-01). "Visualization of podocyte substructure with structured illumination microscopy (SIM): a new approach to nephrotic disease". Biomedical Optics Express. 7 (2): 302–311. ISSN 2156-7085. PMC 4771450 . PMID 26977341. doi:10.1364/BOE.7.000302.
- ↑ Liu, James; Wang, Mei; Tulman, David; Mandava, Sree H.; Elfer, Katherine N.; Gabrielson, Andrew; Lai, Weil; Abshire, Caleb; Sholl, Andrew B. (2016). "Nondestructive Diagnosis of Kidney Cancer on 18-gauge Core Needle Renal Biopsy Using Dual-color Fluorescence Structured Illumination Microscopy". Urology. 98: 195–199. PMID 27597632. doi:10.1016/j.urology.2016.08.036.
- ↑ Westmoreland, David (2016). "Super-resolution microscopy as a potential approach to diagnosis of platelet granule disorders.". Journal of Thrombosis and Haemostasis. 14: 839–849.
- 1 2 3 Reymann, J; Baddeley, D; Gunkel, M; Lemmer, P; Stadter, W; Jegou, T; Rippe, K; Cremer, C; Birk, U (2008). "High-precision structural analysis of subnuclear complexes in fixed and live cells via spatially modulated illumination (SMI) microscopy". Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 16 (3): 367–82. PMID 18461478. doi:10.1007/s10577-008-1238-2.
- ↑ Heintzmann, R.; Cremer, C. (1999). "Lateral modulated excitation microscopy: Improvement of resolution by using a diffraction grating". Proc. SPIE. Optical Biopsies and Microscopic Techniques III. 3568: 185–196. doi:10.1117/12.336833.
- ↑ US patent 7,342,717, filed 10 July 1997: Christoph Cremer, Michael Hausmann, Joachim Bradl, Bernhard Schneider Wave field microscope with detection point spread function
- ↑ Best, G; Amberger, R; Baddeley, D; Ach, T; Dithmar, S; Heintzmann, R; Cremer, C (2011). "Structured illumination microscopy of autofluorescent aggregations in human tissue". Micron. 42 (4): 330–335. doi:10.1016/j.micron.2010.06.016.
- ↑ Stefan W. Hell; Jan Wichmann (1994). "Breaking the diffraction resolution limit by stimulated emission: stimulated-emission-depletion fluorescence microscopy". Optics Letters. 19 (11): 780–2. Bibcode:1994OptL...19..780H. PMID 19844443. doi:10.1364/OL.19.000780.
- ↑ Thomas A. Klar; Stefan W. Hell (1999). "Subdiffraction resolution in far-field fluorescence microscopy". Optics Letters. 24 (14): 954–956. Bibcode:1999OptL...24..954K. PMID 18073907. doi:10.1364/OL.24.000954.
- ↑ Kwon, Jiwoong; Youngbin Lim; Jiwon Jung; Seong Keun Kim (2012). "New sub-diffraction-limit microscopy technique: Dual-point illumination AND-gate microscopy on nanodiamonds (DIAMOND)". Optics Express. 20 (12): 13347–13356. Bibcode:2012OExpr..2013347K. PMID 22714363. doi:10.1364/OE.20.013347. Retrieved 22 October 2013.
- ↑ Fernandes-Suares, M.; Ting, A. Y. (2008). "Fluorescent probes for super-resolution imaging in living cells". Nature Reviews Molecular Cell Biology. 9 (12): 929–943. PMID 19002208. doi:10.1038/nrm2531.
- ↑ Hell, S. W.; et al. (2007). "Far-Field Optical Nanoscopy". Science. 316 (5828): 1153–1158. Bibcode:2007Sci...316.1153H. PMID 17525330. doi:10.1126/science.1137395.
- ↑ Huang, Bo; Bates, M.; Zhuang, X. (2009). "Super resolution fluorescence microscopy". Annual Review of Biochemistry. 78: 993–1016. PMC 2835776 . PMID 19489737. doi:10.1146/annurev.biochem.77.061906.092014.
- ↑ Willig, C.; et al. (2006). "STED microscopy reveals that synaptotagmin remains clustered after synaptic vesicle exocytosis". Nature Letters. 440 (7086): 935–939. Bibcode:2006Natur.440..935W. PMID 16612384. doi:10.1038/nature04592.
- ↑ Patterson, G. H. (2009). "Fluorescence microscopy below the diffraction limit". Seminars in Cell & Developmental Biology. 20 (8): 886–893. PMC 2784032 . PMID 19698798. doi:10.1016/j.semcdb.2009.08.006.
- ↑ Westphal, V.; et al. (2007). "Dynamic far-field nanoscopy". New Journal of Physics. 9 (12): 435. Bibcode:2007NJPh....9..435W. doi:10.1088/1367-2630/9/12/435.
- ↑ Westphal, V.; et al. (2008). "Video-Rate Far-FieldOptical Nanoscopy Dissects Synaptic Vesicle Movement". Science. 320 (5873): 246–249. Bibcode:2008Sci...320..246W. PMID 18292304. doi:10.1126/science.1154228.
- ↑ Chmyrov, Andriy; et al. (2008). "Characterization of new fluorescent labels for ultra-high resolution microscopy". Photochemical & photobiological sciences. 7 (11): 1378–1385. PMID 18958325. doi:10.1039/B810991P.
- ↑ Gustafsson, M. G. L. (2005). "Nonlinear structured-illumination microscopy: Wide-field fluorescence imaging with theoretically unlimited resolution". Proceedings of the National Academy of Sciences. 102 (37): 13081–13086. Bibcode:2005PNAS..10213081G. PMC 1201569 . PMID 16141335. doi:10.1073/pnas.0406877102.
- 1 2 Weisenburger, Siegfried; Boening, Daniel; Schomburg, Benjamin; Giller, Karin; Becker, Stefan; Griesinger, Christian; Sandoghdar, Vahid (2017). "Cryogenic optical localization provides 3D protein structure data with Angstrom resolution". Nature Methods. 14 (2): 141–144. PMID 28068317. doi:10.1038/nmeth.4141.
- 1 2 3 Lemmer P, Gunkel M, Baddeley D, Kaufmann R, Urich A, Weiland Y, Reymann J, Müller P, Hausmann M, Cremer C (2008). "SPDM: Light Microscopy with Single Molecule Resolution at the Nanoscale" (PDF). Applied Physics B. 93: 1–12. Bibcode:2008ApPhB..93....1L. doi:10.1007/s00340-008-3152-x.
- ↑ A.M. van Oijen; J Köhler; J Schmidt; M Müller; G. J Brakenhoff (1998-07-31). "3-Dimensional super-resolution by spectrally selective imaging". Chemical Physics Letters. 292 (1–2): 183–187. Bibcode:1998CPL...292..183V. doi:10.1016/S0009-2614(98)00673-3.
- ↑ Bornfleth Sätzler; Eils Cremer (1998-02-01). "High-precision distance measurements and volume-conserving segmentation of objects near and below the resolution limit in three-dimensional confocal fluorescence microscopy". Journal of Microscopy. 189 (2): 118–136. doi:10.1046/j.1365-2818.1998.00276.x.
- ↑ Reymann, J; Baddeley, D; Gunkel, M; Lemmer, P; Stadter, W; Jegou, T; Rippe, K; Cremer, C; Birk, U (May 2008). "High-precision structural analysis of subnuclear complexes in fixed and live cells via spatially modulated illumination (SMI) microscopy". Chromosome Research: an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology. 16 (3): 367–82. PMID 18461478. doi:10.1007/s10577-008-1238-2.
- ↑ Cremer, R. Kaufmann, M. Gunkel, S. Pres, Y. Weiland, P- Müller, T. Ruckelshausen, P. Lemmer, F. Geiger, S. Degenhard, C. Wege, N-A. W. Lemmermann, R. Holtappels, H. Strickfaden, M. Hausmann (2011): Superresolution imaging of biological nanostructures by spectral precision distance microscopy: Biotechnology 6: 1037–1051
- ↑ C. Cremer, R.Kaufmann, M. Gunkel, F. Polanski, P. Müller, R. Dierkes, S. Degenhard, C. Wege, M. Hausmann, U. Birk:"Application perspectives of localization microscopy in virology", Histochem Cell Biol (2014)
- ↑ Qiaoyun Wang, Rüdiger Dierkes, Rainer Kaufmanna, Christoph Cremer: "Quantitative analysis of individual hepatocyte growth factor receptor clusters in influenza A virus infected human epithelial cells using localization microscopy", Biochimica et Biophysica Acta (2014)
- ↑ Manuel Gunkel, Fabian Erdel, Karsten Rippe, Paul Lemmer, Rainer Kaufmann, Christoph Hörmann, Roman Amberger and Christoph Cremer (2009): Dual color localization microscopy of cellular nanostructures. In: Biotechnology Journal, 2009, 4, 927–938. ISSN 1860-6768
- ↑ Zondervan, R.; Kulzer, F.; Kolchenko, M.; Orrit, M. (2004). "Photobleaching of Rhodamine 6G in Poly(vinyl alcohol) at the Ensemble and Single-Molecule Levels". J. Phys. Chem. A. 108 (10): 1657–1665. doi:10.1021/jp037222e.
- ↑ Weisenburger, Siegfried; Jing, Bo; Renn, Alois; Sandoghdar, Vahid (2013). "Cryogenic localization of single molecules with angstrom precision". Proc. SPIE. Nanoimaging and Nanospectroscopy. 8815: 88150D. doi:10.1117/12.2025373.
- ↑ Imaging chromatin nanostructure with binding-activated localization microscopy based on DNA structure fluctuations Aleksander Szczurek, Ludger Klewes, Jun Xing, Amine Gourram, Udo Birk, Hans Knecht, Jurek W. Dobrucki, Sabine Mai, Christoph Cremer. Nucl Acids Res (2017) Published: 13 January 2017 DOI: https://doi.org/10.1093/nar/gkw1301
- 1 2 Rust, M.; M. Bates; X. Zhuang (2006). "Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM)". Nature Methods. 3 (10): 793–796. doi:10.1038/nmeth929.
- 1 2 3 Betzig, E.; et al. (2006). "Imaging Intracellular Fluorescent Proteins at Nanometer Resolution". Science. 313 (5793): 1642–1645. Bibcode:2006Sci...313.1642B. PMID 16902090. doi:10.1126/science.1127344.
- 1 2 3 Hess, S.; T. Giririjan; M. Mason (2006). "Ultra-High Resolution Imaging by Fluorescence Photoactivation Localization Microscopy". Biophysical Journal. 91 (11): 4258–4272. Bibcode:2006BpJ....91.4258H. PMC 1635685 . PMID 16980368. doi:10.1529/biophysj.106.091116.
- ↑ Zhuang, X. (2009). "Nano-imaging with Storm". Nature Photonics. 3 (7): 365–367. Bibcode:2009NaPho...3..365Z. PMC 2840648 . PMID 20300445. doi:10.1038/nphoton.2009.101.
- ↑ Bates, M.; T. Blosser; X. Zhuang (2005). "Short-range spectroscopic ruler based on a single-molecule optical switch". Physical Review Letters. 94 (108101): 108101. Bibcode:2005PhRvL..94j8101B. PMC 2652517 . PMID 15783528. arXiv:q-bio/0502012 . doi:10.1103/physrevlett.94.108101.
- ↑ Dempsey, G.; M. Bates; W. Kowtoniuk; D.Liu; R. Tsien; X. Zhuang (2009). "Photoswitching Mechanism of Cyanine Dyes". Journal of the American Chemical Society. 131 (51): 18192–18193. PMC 2797371 . PMID 19961226. doi:10.1021/ja904588g.
- 1 2 Bock, H.; et al. (2007). "Two-color far-field fluorescence nanoscopy based on photoswitchable emitters". Applied Physics B. 88 (2): 161–165. Bibcode:2007ApPhB..88..161B. doi:10.1007/s00340-007-2729-0.
- ↑ Folling, J.; et al. (2008). "Fluorescence nanoscopy by ground-state depletion and single-molecule return". Nature Methods. 5 (11): 943–945. PMID 18794861. doi:10.1038/nmeth.1257.
- ↑ Heilemann, M.; et al. (2008). "Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes". Angewandte Chemie International Edition. 47 (33): 6172–6176. PMID 18646237. doi:10.1002/anie.200802376.
- ↑ Heilemann, M.; S. van de Linde; A. Mukherjee; M. Sauer (2009). "Super-resolution imaging with small organic fluorophores". Angewandte Chemie International Edition. 48 (37): 6903–6908. doi:10.1002/anie.200902073.
- ↑ Vogelsang, J.; T. Cordes; C. Forthmann; C. Steinhauer; P. Tinnefeld (2009). "Controlling the fluorescence of ordinary oxazine dyes for single-molecule switching and superresolution microscopy". Proceedings of the National Academy of Sciences. 106 (20): 8107–8112. Bibcode:2009PNAS..106.8107V. PMC 2688868 . PMID 19433792. doi:10.1073/pnas.0811875106.
- ↑ Lee, H.; et al. (2010). "Superresolution Imaging of Targeted Proteins in Fixed and Living Cells Using Photoactivatable Organic Fluorophores". Journal of the American Chemical Society. 132 (43): 15099–15101. PMC 2972741 . PMID 20936809. doi:10.1021/ja1044192.
- 1 2 3 Bates, M.; B. Huang; G. Dempsey; X. Zhuang (2007). "Multicolor Super-resolution Imaging with Photo-switchable Fluorescent Probes". Science. 317 (5845): 1749–1753. Bibcode:2007Sci...317.1749B. PMC 2633025 . PMID 17702910. doi:10.1126/science.1146598.
- 1 2 3 4 Huang, B.; S. Jones; B. Brandenburg; X. Zhuang (2008). "Whole cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution". Nature Methods. 5 (12): 1047–1052. PMC 2596623 . PMID 19029906. doi:10.1038/nmeth.1274.
- 1 2 Dani, A.; B. Huang; J. Bergan; C. Dulac; X. Zhuang (2010). "Super-resolution Imaging of Chemical Synapses in the Brain". Neuron. 68 (5): 843–856. PMC 3057101 . PMID 21144999. doi:10.1016/j.neuron.2010.11.021.
- ↑ Testa, I.; et al. (2010). "Multicolor Fluorescence Nanoscopy in Fixed and Living Cells by Exciting Conventional Fluorophores with a Single Wavelength". Biophysical Journal. 99 (8): 2686–2694. Bibcode:2010BpJ....99.2686T. PMC 2956215 . PMID 20959110. doi:10.1016/j.bpj.2010.08.012.
- 1 2 3 4 Jones, S.; S. Shim; X. Zhuang (2011). "Fast three-dimensional super-resolution imaging of live cells". Nature Methods. 8 (6): 499–508. PMC 3137767 . PMID 21552254. doi:10.1038/nmeth.1605.
- 1 2 Wang, W.; G. Li; C. Chen; S. Xie; X. Zhuang (2011). "Chromosome organization by a nucleoid-associated protein in live bacteria". Science. 333 (6048): 1445–1449. Bibcode:2011Sci...333.1445W. PMC 3329943 . PMID 21903814. doi:10.1126/science.1204697.
- 1 2 Huang, B.; W. Wang; M. Bates; X. Zhuang (2008). "Three-dimensional Super-resolution Imaging by Stochastic Optical Reconstruction Microscopy". Science. 319 (5864): 810–813. Bibcode:2008Sci...319..810H. PMC 2633023 . PMID 18174397. doi:10.1126/science.1153529.
- ↑ Wu, M.; B. Huang; M. Graham; A. Raimondi; J. Heuser; X. Zhuang; P. Camilli (2010). "Coupling between clathrin-dependent endocytic budding and F-BAR-dependent tubulation in a cell-free system". Nature Cell Biology. 12 (9): 902–908. PMC 3338250 . PMID 20729836. doi:10.1038/ncb2094.
- ↑ Kaufmann, Rainer; Müller, Patrick; Hausmann, Michael; Cremer, Christoph (2010). "Imaging label-free intracellular structures by localisation microscopy". Micron. doi:10.1016/j.micron.2010.03.06 (inactive 2017-07-23).
- ↑ Ayas, S. (2013). "Label-Free Nanometer-Resolution Imaging of Biological Architectures through Surface Enhanced Raman Scattering". Scientific Reports. 3: 2624. Bibcode:2013NatSR...3E2624A. doi:10.1038/srep02624.
- ↑ Heilemann et al, 2008, Angewandte Chemie and van de Linde et al, 2011, Photochem. Photobiol. Sci
- ↑ Baddeley, David; Jayasinghe, Isuru D.; Cremer, Christoph; Cannell, Mark B.; Soeller, Christian (January 2009). "Light-Induced Dark States of Organic Fluochromes Enable 30 nm Resolution Imaging in Standard Media". Biophysical Journal. 96 (2): L22–L24. Bibcode:2009BpJ....96L..22B. PMC 2716455 . PMID 19167284. doi:10.1016/j.bpj.2008.11.002.
- ↑ Fölling, Jonas; Bossi, Mariano; Bock, Hannes; Medda, Rebecca; Wurm, Christian A; Hein, Birka; Jakobs, Stefan; Eggeling, Christian; Hell, Stefan W (15 September 2008). "Fluorescence nanoscopy by ground-state depletion and single-molecule return". Nature Methods. 5 (11): 943–945. PMID 18794861. doi:10.1038/NMETH.1257.
- ↑ Heilemann, M; van de Linde, S; Schüttpelz, M; Kasper, R; Seefeldt, B; Mukherjee, A; Tinnefeld, P; Sauer, M (2008). "Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes.". Angewandte Chemie International Edition in English. 47 (33): 6172–6. PMID 18646237. doi:10.1002/anie.200802376.
- ↑ Sage, Daniel; Kirshner, Hagai; Pengo, Thomas; Stuurman, Nico; Min, Junhong; Manley, Suliana; Unser, Michael (15 June 2015). "Quantitative evaluation of software packages for single-molecule localization microscopy". 12 (8): 717–724. doi:10.1038/nmeth.3442.
- ↑ Baddeley, David; Cannell, Mark B.; Soeller, Christian (18 January 2010). "Visualization of Localization Microscopy Data". Microscopy and Microanalysis. 16 (1): 64–72. PMID 20082730. doi:10.1017/S143192760999122X.
- ↑ Baddeley D, Batram C, Weiland Y, Cremer C, Birk UJ.: Nanostructure analysis using Spatially Modulated Illumination microscopy. In: Nature Protocols 2007; 2: 2640–2646
- ↑ Rainer Kaufmann, Patrick Müller, Georg Hildenbrand, Michael Hausmann & Christoph Cremer (2010): Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy, Journal of Microscopy 2010, doi:10.1111/j.1365-2818.2010.03436.x PMID 21118230
- ↑ Liss, Viktoria; et al. (2015). "Self-labelling enzymes as universal tags for fluorescence microscopy, super-resolution microscopy and electron microscopy". Scientific Reports. 5. doi:10.1038/srep17740.
Further reading
- Marx, Vivien (26 November 2013). "Is super-resolution microscopy right for you?". Technology Feature. Nature Methods (Paper "Nature Reprint Collection, Technology Features" (Nov 2014)). 10 (12): 1157–63. PMID 24296472. doi:10.1038/nmeth.2756. (Registration required (help)).
- Cremer, Christoph; Masters, Barry R. (2013). "Resolution enhancement techniques in microscopy". The European Physical Journal H. 38 (3): 281. doi:10.1140/epjh/e2012-20060-1.
Optical microscopy | ||
|---|---|---|
| Illumination and contrast methods |  | |
| Fluorescence methods | ||
| Sub-diffraction limit techniques | ||