Simplified molecular-input line-entry system
| Filename extension |
.smi |
|---|---|
| Internet media type |
chemical/x-daylight-smiles |
| Type of format | chemical file format |
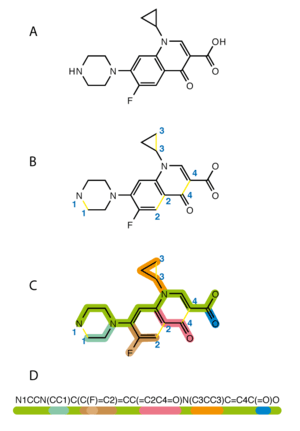
The simplified molecular-input line-entry system (SMILES) is a specification in form of a line notation for describing the structure of chemical species using short ASCII strings. SMILES strings can be imported by most molecule editors for conversion back into two-dimensional drawings or three-dimensional models of the molecules.
The original SMILES specification was initiated in the 1980s. It has since been modified and extended. In 2007, an open standard called "OpenSMILES" was developed in the open-source chemistry community. Other 'linear' notations include the Wiswesser Line Notation (WLN), ROSDAL and SLN.
History
The original SMILES specification was initiated by David Weininger at the USEPA Mid-Continent Ecology Division Laboratory in Duluth in the 1980s.[1][2][3][4] Acknowledged for their parts in the early development were "Gilman Veith and Rose Russo (USEPA) and Albert Leo and Corwin Hansch (Pomona College) for supporting the work, and Arthur Weininger (Pomona; Daylight CIS) and Jeremy Scofield (Cedar River Software, Renton, WA) for assistance in programming the system."[5] The Environmental Protection Agency funded the initial project to develop SMILES.[6][7]
It has since been modified and extended by others, most notably by Daylight Chemical Information Systems. In 2007, an open standard called "OpenSMILES" was developed by the Blue Obelisk open-source chemistry community. Other 'linear' notations include the Wiswesser Line Notation (WLN), ROSDAL and SLN (Tripos Inc).
In July 2006, the IUPAC introduced the InChI as a standard for formula representation. SMILES is generally considered to have the advantage of being slightly more human-readable than InChI; it also has a wide base of software support with extensive theoretical (e.g., graph theory) backing.
Terminology
The term SMILES refers to a line notation for encoding molecular structures and specific instances should strictly be called SMILES strings. However, the term SMILES is also commonly used to refer to both a single SMILES string and a number of SMILES strings; the exact meaning is usually apparent from the context. The terms "canonical" and "isomeric" can lead to some confusion when applied to SMILES. The terms describe different attributes of SMILES strings and are not mutually exclusive.
Typically, a number of equally valid SMILES strings can be written for a molecule. For example, CCO, OCC and C(O)C all specify the structure of ethanol. Algorithms have been developed to generate the same SMILES string for a given molecule; of the many possible strings, these algorithms choose only one of them. This SMILES is unique for each structure, although dependent on the canonicalization algorithm used to generate it, and is termed the canonical SMILES. These algorithms first convert the SMILES to an internal representation of the molecular structure; an algorithm then examines that structure and produces a unique SMILES string. Various algorithms for generating canonical SMILES have been developed and include those by Daylight Chemical Information Systems, OpenEye Scientific Software, MEDIT, Chemical Computing Group, MolSoft LLC, and the Chemistry Development Kit. A common application of canonical SMILES is indexing and ensuring uniqueness of molecules in a database.
The original paper that described the CANGEN[2] algorithm claimed to generate unique SMILES strings for graphs representing molecules, but the algorithm fails for a number of simple cases (e.g. cuneane, 1,2-dicyclopropylethane) and cannot be considered a correct method for representing a graph canonically.[8] There is currently no systematic comparison across commercial software to test if such flaws exist in those packages.
SMILES notation allows the specification of configuration at tetrahedral centers, and double bond geometry. These are structural features that cannot be specified by connectivity alone and SMILES which encode this information are termed isomeric SMILES. A notable feature of these rules is that they allow rigorous partial specification of chirality. The term isomeric SMILES is also applied to SMILES in which isotopes are specified.
Graph-based definition
In terms of a graph-based computational procedure, SMILES is a string obtained by printing the symbol nodes encountered in a depth-first tree traversal of a chemical graph. The chemical graph is first trimmed to remove hydrogen atoms and cycles are broken to turn it into a spanning tree. Where cycles have been broken, numeric suffix labels are included to indicate the connected nodes. Parentheses are used to indicate points of branching on the tree.
The resultant SMILES form depends on the choices:
- of the bonds chosen to break cycles,
- of the starting atom used for the depth-first traversal, and
- of the order in which branches are listed when encountered.
Description
Atoms
Atoms are represented by the standard abbreviation of the chemical elements, in square brackets, such as [Au] for gold. Brackets may be omitted in the common case of atoms which:
- are in the "organic subset" of B, C, N, O, P, S, F, Cl, Br, or I, and
- have no formal charge, and
- have the number of hydrogens attached implied by their normal valence, and
- are the normal isotopes, and
- are not chiral centers.
All other elements must be enclosed in brackets, and have charges and hydrogens shown explicitly. For instance, the SMILES for water may be written as either O or [OH2]. Hydrogen may also be written as a separate atom; water may also be written as [H]O[H].
When brackets are used, the symbol H is added if the atom in brackets is bonded to one or more hydrogen, followed by the number of hydrogen atoms if greater than 1, then by the sign '+' for a positive charge or by '-' for a negative charge. For example, [NH4+] for ammonium. If there is more than one charge, it is normally written as digit; however, it is also possible to repeat the sign as many times as the ion has charges: one may write either [Ti+4] or [Ti++++] for Titanium IV (Ti4+). Thus, the hydroxide anion is represented by [OH-], the hydronium cation is [OH3+] and the cobalt III cation (Co3+) is either [Co+3] or [Co+++].
Bonds
A bond is represented using one of the symbols '.' '-' '=' '#' '$' ':' '/' or '\'.
Bonds between aliphatic atoms are assumed to be single unless specified otherwise and are implied by adjacency in the SMILES string. Although single bonds may be written as "-", this is usually omitted. For example, the SMILES for ethanol may be written as C-C-O, CC-O or C-CO, but is usually written CCO.
Double, triple, and quadruple bonds are represented by the symbols '=', '#', and '$' respectively as illustrated by the SMILES O=C=O (carbon dioxide), C#N (hydrogen cyanide) and [Ga-]$[As+] (gallium arsenide).
An additional type of bond is a "non-bond", indicated with ".", to indicate that two parts are not bonded together. For example, aqueous sodium chloride may be written as [Na+].[Cl-] to show the dissociation.
An aromatic "one and a half" bond may be indicated with ':'; see § Aromaticity below.
Single bonds adjacent to double bonds may be represented using '/' or '\' to indicate stereochemical configuration; see § Stereochemistry below.
Rings
Ring structures are written by breaking each ring at an arbitrary point (although some choices will lead to a more legible SMILES than others) to make an acyclic structure and adding numerical ring closure labels to show connectivity between non-adjacent atoms.
For example, cyclohexane and dioxane may be written as C1CCCCC1 and O1CCOCC1 respectively. For a second ring, the label will be 2. For example, decalin (decahydronaphthalene) may be written as C1CCCC2C1CCCC2.
SMILES does not require that ring numbers be used in any particular order, and permits ring number zero, although this is rarely used. Also, it is permitted to re-use ring numbers after the first ring has closed, although this usually makes formulae harder to read. For example, bicyclohexyl is usually written as C1CCCCC1C2CCCCC2, but it may also be written as C0CCCCC0C0CCCCC0.
Multiple digits after a single atom indicate multiple ring-closing bonds. For example, an alternative SMILES notation for decalin is C1CCCC2CCCCC12, where the final carbon participates in both ring-closing bonds 1 and 2. If two-digit ring numbers are required, the label is preceded by %, so "C%12" is a single ring-closing bond, of ring 12.
Ring-closing digits may be preceded by a bond type. For example, cyclopropene is usually written C1=CC1, but if the double bond is chosen as the ring-closing bond, it may be written as C=1CC1, C1CC=1, or C=1CC=1. (The first form is preferred.) C=1CC-1 is illegal, as it explicitly specifies conflicting types for the ring-closing bond.
Ring-closing bonds may not be used to denote multiple bonds. For example, C1C1 is not a valid alternative to C=C for ethylene. However, they may be used with non-bonds; C1.C2.C12 is a peculiar but legal alternative way to write propane, more commonly written CCC.
Choosing a ring-break point adjacent to attached groups can lead to a simpler SMILES form by avoiding branches. For example, cyclohexane-1,2-diol is most simply written as OC1CCCCC1O; choosing a different ring-break location produces a branched structure that requires parentheses to write.
Aromaticity
Aromatic rings such as benzene may be written in one of three forms:
- In Kekulé form with alternating single and double bonds, e.g. C1=CC=CC=C1,
- Using the aromatic bond symbol ":", e.g. C:1:C:C:C:C:C1, or
- Most commonly, by writing the constituent B, C, N, O, P and S atoms in lower-case forms 'b', 'c', 'n', 'o', 'p' and 's', respectively.
In the latter case, bonds between two aromatic atoms are assumed (if not explicitly shown) to be aromatic bonds. Thus, benzene, pyridine and furan can be represented respectively by the SMILES c1ccccc1, n1ccccc1 and o1cccc1.
Aromatic nitrogen bonded to hydrogen, as found in pyrrole must be represented as [nH] and imidazole is written in SMILES notation as n1c[nH]cc1.
When aromatic atoms are singly bonded to each other, such as in biphenyl, a single bond must be shown explicitly: c1ccccc1-c2ccccc2. This is one of the few cases where the single bond symbol "-" is required. (In fact, most SMILES software can correctly infer that the bond between the two rings cannot be aromatic and so will accept the form "c1ccccc1c2ccccc2".)
The Daylight and OpenEye algorithms for generating canonical SMILES differ in their treatment of aromaticity.

Branching
Branches are described with parentheses, as in CCC(=O)O for propionic acid and FC(F)F for fluoroform. The first atom within the parentheses, and the first atom after the parenthesized group, are both bonded to the same branch point atom.
Substituted rings can be written with the branching point in the ring as illustrated by the SMILES COc(c1)cccc1C#N (see depiction) and COc(cc1)ccc1C#N (see depiction) which encode the 3 and 4-cyanoanisole isomers. Writing SMILES for substituted rings in this way can make them more human-readable.
Branches may be written in any order. For example, bromochlorodifluoromethane may be written as FC(Br)(Cl)F, BrC(F)(F)Cl, C(F)(Cl)(F)Br, or the like. Generally, a SMILES form is easiest to read if the simpler branch comes first, with the final, unparenthesized portion being the most complex. The only caveats to such rearrangements are:
- If ring numbers are reused, they are paired according to their order of appearance in the SMILES string. Some adjustments may be required to preserve the correct pairing.
- If stereochemistry is specified, adjustments must be made; see Stereochemistry § Notes below.
The one form of branch which does not require parentheses are ring-closing bonds. Choosing ring-closing bonds appropriately can reduce the number of parentheses required. For example, toluene is normally written as Cc1ccccc1 or c1ccccc1C, avoiding the parentheses required if written as c1ccc(C)ccc1 or c1ccc(ccc1)C.
Stereochemistry
SMILES permits, but does not require, specification of stereoisomers.
Configuration around double bonds is specified using the characters "/" and "\" to show directional single bonds adjacent to a double bond. For example, F/C=C/F (see depiction) is one representation of trans-difluoroethene, in which the fluorine atoms are on opposite sides of the double bond, whereas F/C=C\F (see depiction) is one possible representation of cis-difluoroethene, in which the Fs are on the same side of the double bond, as shown in the figure.
Configuration at tetrahedral carbon is specified by @ or @@. L-Alanine, the more common enantiomer of the amino acid alanine can be written as N[C@@H](C)C(=O)O (see depiction). The @@ specifier indicates that, when viewed from nitrogen along the bond to the chiral center, the sequence of substituents hydrogen (H), methyl (C) and carboxylate (C(=O)O) appear clockwise. D-Alanine can be written as N[C@H](C)C(=O)O (see depiction). For the most part, the order in which branches are specified is unimportant, but in this case it is important. If the branches are reversed to alanine is written as NC(C(=O)O)C, then the configuration also reverses; L-alanine is written as N[C@H](C(=O)O)C (see depiction).
Isotopes
Isotopes are specified with a number equal to the integer isotopic mass preceding the atomic symbol. Benzene in which one atom is carbon-14 is written as [14c]1ccccc1 and deuterochloroform is [2H]C(Cl)(Cl)Cl.
Examples
| Molecule | Structure | SMILES Formula |
|---|---|---|
| Dinitrogen | N≡N | N#N |
| Methyl isocyanate (MIC) | CH3–N=C=O | CN=C=O |
| Copper(II) sulfate | Cu2+ SO42− | [Cu+2].[O-]S(=O)(=O)[O-] |
| Vanillin |  |
O=Cc1ccc(O)c(OC)c1 OCc1cc(C=O)ccc1O |
| Melatonin (C13H16N2O2) |  |
CC(=O)NCCC1=CNc2c1cc(OC)cc2 CC(=O)NCCc1c[nH]c2ccc(OC)cc12 |
| Flavopereirin (C17H15N2) |  |
CCc(c1)ccc2[n+]1ccc3c2[nH]c4c3cccc4 CCc1c[n+]2ccc3c4ccccc4[nH]c3c2cc1 |
| Nicotine (C10H14N2) |  |
CN1CCC[C@H]1c2cccnc2 |
| Oenanthotoxin (C17H22O2) |  |
CCC[C@@H](O)CC\C=C\C=C\C#CC#C\C=C\CO CCC[C@@H](O)CC/C=C/C=C/C#CC#C/C=C/CO |
| Pyrethrin II (C22H28O5) |  |
COC(=O)/C(C)=C/C1C(C)(C)[C@H]1C(=O)O[C@H]2CC(=O)C(=C2C)C/C=C/C=C |
| Aflatoxin B1 (C17H12O6) |  |
O1C=C[C@H]([C@H]1O2)c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5 |
| Glucose (glucopyranose) (C6H12O6) |  |
OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)1 |
| Bergenin (cuscutin) (a resin) (C14H16O9) |  |
OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H]2[C@@H]1c3c(O)c(OC)c(O)cc3C(=O)O2 |
| A pheromone of the Californian scale insect |  |
CC(=O)OCCC(/C)=C\C[C@H](C(C)=C)CCC=C |
| 2S,5R-Chalcogran: a pheromone of the bark beetle Pityogenes chalcographus[9] |  |
CC[C@H](O1)CC[C@@]12CCCO2 |
| Alpha-thujone (C10H16O) |  |
CC(C)[C@@]12C[C@@H]1[C@@H](C)C(=O)C2 |
| Thiamine (C12H17N4OS+) (vitamin B1) |
 |
OCCc1c(C)[n+](cs1)Cc2cnc(C)nc2N |
To illustrate a molecule with more than 9 rings, consider Cephalostatin-1,[10] a steroidic trisdecacyclic pyrazine with the empirical formula C54H74N2O10 isolated from the Indian Ocean hemichordate Cephalodiscus gilchristi:

Starting with the left-most methyl group in the figure:
CC(C)(O1)C[C@@H](O)[C@@]1(O2)[C@@H](C)[C@@H]3CC=C4[C@]3(C2)C(=O)C[C@H]5[C@H]4CC[C@@H](C6)[C@]5(C)Cc(n7)c6nc(C[C@@]89(C))c7C[C@@H]8CC[C@@H]%10[C@@H]9C[C@@H](O)[C@@]%11(C)C%10=C[C@H](O%12)[C@]%11(O)[C@H](C)[C@]%12(O%13)[C@H](O)C[C@@]%13(C)CO
Note that '%' appears in front of the index of ring closure labels above 9; see § Rings above.
Other examples of SMILES
The SMILES notation is described extensively in the SMILES theory manual provided by Daylight Chemical Information Systems and a number of illustrative examples are presented. Daylight's depict utility provides users with the means to check their own examples of SMILES and is a valuable educational tool.
Extensions
SMARTS is a line notation for specification of substructural patterns in molecules. While it uses many of the same symbols as SMILES, it also allows specification of wildcard atoms and bonds, which can be used to define substructural queries for chemical database searching. One common misconception is that SMARTS-based substructural searching involves matching of SMILES and SMARTS strings. In fact, both SMILES and SMARTS strings are first converted to internal graph representations which are searched for subgraph isomorphism. SMIRKS is a line notation for specifying reaction transforms.
Conversion
SMILES can be converted back to 2-dimensional representations using Structure Diagram Generation algorithms (Helson, 1999). This conversion is not always unambiguous. Conversion to 3-dimensional representation is achieved by energy minimization approaches. There are many downloadable and web-based conversion utilities.
See also
- SMILES arbitrary target specification SMARTS language for specification of substructural queries.
- SYBYL Line Notation (another line notation)
- Molecular Query Language – query language allowing also numerical properties, e.g. physicochemical values or distances
- Chemistry Development Kit (2D layout and conversion)
- International Chemical Identifier (InChI), the IUPAC's alternative to SMILES.
- OpenBabel, JOELib, OELib (conversion)
References
- ↑ Weininger 1988
- 1 2 Weininger, Weininger & Weininger 1989
- ↑ Weininger 1990
- ↑ Swanson, Richard Pommier (2004). "The Entrance of Informatics into Combinatorial Chemistry". In Rayward, W. [Warden] Boyd; Bowden, Mary Ellen. The History and Heritage of Scientific and Technological Information Systems: Proceedings of the 2002 Conference of the American Society of Information Science and Technology and the Chemical Heritage Foundation. Medford, NJ: Information Today. p. 205. ISBN 1-57387-229-6.
https://wayback.archive-it.org/2118/20100925010036/http://64.251.202.97/pubs/asist2002/17-swanson.pdf
- ↑ Weininger, Dave. "Acknowledgements on Daylight Tutorial smiles-etc page". Retrieved 24 June 2013.
- ↑ Anderson, Veith & Weininger 1987
- ↑ "SMILES Tutorial: What is SMILES?". U.S. Environmental Protection Agency. Retrieved 2012-09-23.
- ↑ Hutchison D, Kanade T, Kittler J, Klienberg JM, Mattern F, Mitchell JC, Naor M, Nierstrasz O, Rangan CP, Steffen B, Sudan M, Terzopoulos D, Tygar D, Vardi MY, Weikum G, Raschid L, Neglur G, Grossman RL, Liu B (2005). "Assigning Unique Keys to Chemical Compounds for Data Integration: Some Interesting Counter Examples". In Ludäscher B. Data Integration in the Life Sciences. Lecture Notes in Computer Science. 3615. Berlin: Springer. pp. 145–157. ISBN 978-3-540-27967-9. doi:10.1007/11530084_13. Retrieved 2013-02-12.
- ↑ Byers, JA; Birgersson, G; Löfqvist, J; Appelgren, M; Bergström, G (Mar 1990). "Isolation of pheromone synergists of bark beetle,Pityogenes chalcographus, from complex insect-plant odors by fractionation and subtractive-combination bioassay" (PDF). Journal of Chemical Ecology. 16 (3): 861–76. PMID 24263601. doi:10.1007/BF01016496.
- ↑ National Center for Biotechnology Information (NCBI). PubChem Compound. (accessed May 12, 2012) PubChem Compound CID=183413 (Cephalostatin-1)
Further reading
- Anderson E, Veith GD, Weininger D (1987). SMILES: A line notation and computerized interpreter for chemical structures. Duluth, MN: U.S. EPA, Environmental Research Laboratory-Duluth. Report No. EPA/600/M-87/021.
- Helson HE (1999). "Structure Diagram Generation". In Lipkowitz KB, Boyd DB. Rev. Comput. Chem. 13. New York: Wiley-VCH. pp. 313–398. doi:10.1002/9780470125908.ch6.
- Weininger D (February 1988). "SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules". Journal of Chemical Information and Modeling. 28 (1): 31–6. doi:10.1021/ci00057a005.
- Weininger D, Weininger A, Weininger JL (May 1989). "SMILES. 2. Algorithm for generation of unique SMILES notation". Journal of Chemical Information and Modeling. 29 (2): 97–101. doi:10.1021/ci00062a008.
- Weininger D (August 1990). "SMILES. 3. DEPICT. Graphical depiction of chemical structures". Journal of Chemical Information and Modeling. 30 (3): 237–43. doi:10.1021/ci00067a005.
External links
- "SMILES – A Simplified Chemical Language"
- The OpenSMILES home page
- "SMARTS – SMILES Extension"
- Daylight SMILES tutorial
- Parsing SMILES
SMILES related software utilities
- NCI/CADD Chemical Identifier Resolver – resolves or generates SMILES from chemical names, CAS Registry Numbers, InChI/InChIKey and many other chemical structure file formats
- NCI/CADD Online SMILES Translator and Structure File Generator – Java online molecule editor
- PubChem server side structure editor – online molecule editor
- smi23d – 3D Coordinate Generation
- Daylight Depict – Translate a SMILES formula into graphics
- GIF/PNG-Creator for 2D Plots of Chemical Structures
- JME molecule editor - Chemical editor/viewer and SMILES/SMARTS generator in Java
- JSME molecule editor - Free chemical editor/viewer and SMILES/SMARTS generator in JavaScript
- ACD/ChemSketch
- Marvin by ChemAxon – online chemical editor/viewer and SMILES generator/converter
- Instant JChem by ChemAxon – desktop application for storing/generating/converting/visualizing/searching SMILES structures, particularly batch processing; personal edition free
- JChem for Excel by ChemAxon – MS Excel add-in for storing/generating/converting/visualizing/searching SMILES structures
- Smormo-Ed – a molecule editor for Linux which can read and write SMILES
- InChI.info – an unofficial InChI website featuring on-line converter from InChI and SMILES to molecular drawings
- Balloon – A free program for 3D coordinate generation and conformational analysis.
- Indigo – an open-source cross-platform cheminformatics library with a plugin for IUPAC-compliant molecule and reaction 2D structural formula rendering.
- Open Babel – an open-source chemical toolbox allowing anyone to search, convert, analyze, or store biochemical data.
- Bioclipse – a free and open source workbench for the life sciences
- MolEngine – A .NET cheminformatics toolkit to read/write SMILES, generate 2D coordinate from SMILES, and convert SMILES from/into other Chemical file formats.
- JSDraw – A cross-platform javascript chemical structure editor to generate SMILES and SMARTS.