Selective sweep
In genetics, a selective sweep is the reduction or elimination of variation among the nucleotides near a mutation in DNA. It results from a beneficial allele's having recently reached fixation due to strong positive natural selection.
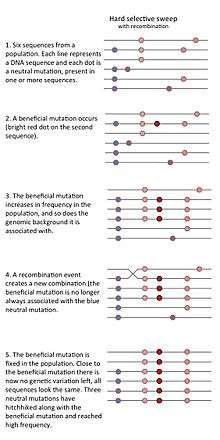
A selective sweep can occur when a rare or previously non-existing allele that increases the fitness of the carrier (relative to other members of the population) increases rapidly in frequency due to natural selection. As the prevalence of such a beneficial allele increases, genetic variants that happen to be present on the genomic background (the DNA neighborhood) of the beneficial allele will also become more prevalent. This phenomenon is called genetic hitchhiking. A selective sweep due to a strongly selected allele, which arose on a single genomic background therefore results in a region of the genome with a large reduction of genetic variation in that chromosome region. The idea that strong positive selection could reduce nearby genetic variation due to hitchhiking was proposed by Maynard-Smith and Haigh in 1974.[1]
It is now recognized that not all sweeps reduce genetic variation in the same way. Sweeps can be categorized in three main categories.
- The "classic selective sweep" or "hard selective sweep" is expected to occur when beneficial mutations are rare, but once a beneficial mutation has occurred it increases in frequency rapidly, thereby drastically reducing genetic variation in the population.[1]
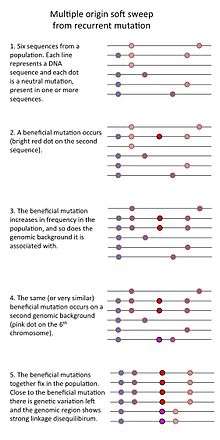
- A so-called "soft sweep from standing genetic variation" occurs when a previously neutral mutation that was present in a population becomes beneficial because of an environmental change. Such a mutation may be present on several genomic backgrounds so that when it rapidly increases in frequency, it doesn't erase all genetic variation in the population.[2]
- Finally, a "multiple origin soft sweep" occurs when mutations are common (for example in a large population) so that the same or similar beneficial mutations occurs on different genomic backgrounds such that no single genomic background can hitchhike to high frequency.[3]

Detection
Whether or not a selective sweep has occurred can be investigated in various ways. One method is to measure linkage disequilibrium, i.e., whether a given haplotype is overrepresented in the population. Under neutral evolution, genetic recombination will result in the reshuffling of the different alleles within a haplotype, and no single haplotype will dominate the population. However, during a selective sweep, selection for a positively selected gene variant will also result in selection of neighbouring alleles and less opportunity for recombination. Therefore, the presence of strong linkage disequilibrium might indicate that there has been a recent selective sweep, and can be used to identify sites recently under selection.
There have been many scans for selective sweeps in humans and other species, using a variety of statistical approaches and assumptions.[4]
In maize, a recent comparison of yellow and white corn genotypes surrounding Y1—the phytoene synthetase gene responsible for the yellow endosperm color, shows strong evidence for a selective sweep in yellow germplasm reducing diversity at this locus and linkage disequilibrium in surrounding regions. White maize lines had increased diversity and no evidence of linkage disequilibrium associated with a selective sweep.[5]
Relevance to disease
Because selective sweeps allow for rapid adaptation, they have been cited as a key factor in the ability of pathogenic bacteria and viruses to attack their hosts and survive the medicines we use to treat them.[6] In such systems, the competition between host and parasite is often characterized as an evolutionary "arms race", so the more rapidly one organism can change its method of attack or defense, the better. This has elsewhere been described by the Red Queen hypothesis. Needless to say, a more effective pathogen or a more resistant host will have an adaptive advantage over its conspecifics, providing the fuel for a selective sweep.
One example comes from the human influenza virus, which has been involved in an adaptive contest with humans for hundreds of years. While antigenic drift (the gradual change of surface antigens) is considered the traditional model for changes in the viral genotype, recent evidence[7] suggests that selective sweeps play an important role as well. In several flu populations, the time to the most recent common ancestor (TMRCA) of "sister" strains, an indication of relatedness, suggested that they had all evolved from a common progenitor within just a few years. Periods of low genetic diversity, presumably resultant from genetic sweeps, gave way to increasing diversity as different strains adapted to their own locales.
A similar case can be found in Toxoplasma gondii, a remarkably potent protozoan parasite capable of infecting warm-blooded animals. Interestingly, T. gondii was recently discovered to exist in only three clonal lineages in all of Europe and North America.[8] In other words, there are only three genetically distinct strains of this parasite in all of the Old World and much of the New World. These three strains are characterized by a single monomorphic version of the gene Chr1a, which emerged at approximately the same time as the three modern clones. It appears then, that a novel genotype emerged containing this form of Chr1a and swept the entire European and North American population of Toxoplasma gondii, bringing with it the rest of its genome via genetic hitchhiking. The South American strains of T. gondii, of which there are far more than exist elsewhere, also carry this allele of Chr1a.
Involvement in agriculture and domestication
Rarely are genetic variability and its opposing forces, including adaptation, more relevant than in the generation of domestic and agricultural species. Cultivated crops, for example, have essentially been genetically modified for more than ten thousand years,[9] subjected to artificial selective pressures, and forced to adapt rapidly to new environments. Selective sweeps provide a baseline from which different varietals could have emerged.[10]
For example, recent study of the corn (Zea mays) genotype uncovered dozens of ancient selective sweeps uniting modern cultivars on the basis of shared genetic data possibly dating back as far as domestic corn's wild counterpart, teosinte. In other words, though artificial selection has shaped the genome of corn into a number of distinctly adapted cultivars, selective sweeps acting early in its development provide a unifying homoplasy of genetic sequence. In a sense, the long-buried sweeps may give evidence of corn's, and teosinte's, ancestral state by elucidating a common genetic background between the two.
Another example of the role of selective sweeps in domestication comes from the chicken. A Swedish research group recently used parallel sequencing techniques to examine eight cultivated varieties of chicken and their closest wild ancestor with the goal of uncovering genetic similarities resultant from selective sweeps.[11] They managed to uncover evidence of several selective sweeps, most notably in the gene responsible for thyroid-stimulating hormone receptor (TSHR), which regulates the metabolic and photoperiod-related elements of reproduction. What this suggests is that, at some point in the domestication of the chicken, a selective sweep, probably driven by human intervention, subtly changed the reproductive machinery of the bird, presumably to the advantage of its human manipulators.
In humans
Examples of selective sweeps in humans are in variants affecting lactase persistence,[12][13] and adaptation to high altitude.[14]
See also
References
- 1 2 Smith, John Maynard; Haigh, John (1974-02-01). "The hitch-hiking effect of a favourable gene". Genetics Research. 23 (1): 23–35. ISSN 1469-5073. PMID 4407212. doi:10.1017/S0016672300014634.
- ↑ Hermisson, Joachim; Pennings, Pleuni S. (2005-04-01). "Soft Sweeps". Genetics. 169 (4): 2335–2352. ISSN 0016-6731. PMC 1449620
 . PMID 15716498. doi:10.1534/genetics.104.036947.
. PMID 15716498. doi:10.1534/genetics.104.036947. - ↑ Pennings, Pleuni S.; Hermisson, Joachim (2006-05-01). "Soft Sweeps II—Molecular Population Genetics of Adaptation from Recurrent Mutation or Migration". Molecular Biology and Evolution. 23 (5): 1076–1084. ISSN 0737-4038. PMID 16520336. doi:10.1093/molbev/msj117.
- ↑ Fu, Wenqing; Akey, Joshua M. (2013). "Selection and adaptation in the human genome". Annual Review of Genomics and Human Genetics. 14: 467–489. ISSN 1545-293X. PMID 23834317. doi:10.1146/annurev-genom-091212-153509.
- ↑ Palaisa K; Morgante M; Tingey S; Rafalski A (June 2004). "Long-range patterns of diversity and linkage disequilibrium surrounding the maize Y1 gene are indicative of an asymmetric selective sweep". Proc. Natl. Acad. Sci. U.S.A. 101 (26): 9885–90. PMC 470768 . PMID 15161968. doi:10.1073/pnas.0307839101.
- ↑ Sa, Juliana Marth, Twua, Olivia Twua, Haytona , Karen, Reyesa, Sahily, Fayb, Michael P., Ringwald, Pascal, & Wellemsa, Thomas E. (2009). "Geographic patterns of Plasmodium falciparum drug resistance distinguished by differential responses to amodiaquine and chloroquine". PNAS. 106 (45): 18883–18889. PMC 2771746 . PMID 19884511. doi:10.1073/pnas.0911317106.
- ↑ Rambaut, Andrew, Pybus, Oliver G., Nelson, Martha I., Viboud, Cecile, Taubenberger, Jeffery K., & Holmes, Edward C. (2008). "The genomic and epidemiological dynamics of human influenza A virus". Nature. 453: 615–619. PMC 2441973 . PMID 18418375. doi:10.1038/nature06945.
- ↑ Sibley, L. David & Ajioka, James W (2008). "Population Structure of Toxoplasma gondii: Clonal Expansion Driven by Infrequent Recombination and Selective Sweeps". Annu. Rev. Microbiol. 62 (1): 329–359. doi:10.1146/annurev.micro.62.081307.162925.
- ↑ Hillman, G., Hedges, R., Moore, A., Colledge, S., & Pettitt, P. (2001). "New evidence of Late glacial cereal cultivation at Abu Hureyra on the Euphrates". Holocene. 4: 388–393.
- ↑ Gore, Michael A., Chia, Jer-Ming, Elshire, Robert J., Sun, Ersoz, Elhan S., Hurwitz, Bonnie L., Peiffer, Jason A., McMullen, Michael D., Grills, George S., Ross-Ibarra, Jeffrey, Ware, Doreen H., & Buckler, Edward S. (2009). "A First-Generation Haplotype Map of Maize". Science. 326: 1115–7. PMID 19965431. doi:10.1126/science.1177837.
- ↑ Rubin, Carl-Johan, Zody, Michael C., Eriksson, Jonas, Meadows, Jennifer R. S., Sherwood, Ellen, Webster, Matthew T., Jiang, Lin, Ingman, Max, Sharpe, Sojeong, Ted Ka, Hallboök, Finn, Besnier, Francois, Carlborg, Orjan, Bed'hom, Bertrand, Tixier-Boichard, Michele, Jensen, Per, Siege, Paul, Lindblad-Toh, Kerstin, & Andersson, Leif (March 2010). "Whole-genome resequencing reveals loci under selection during chicken domestication". Letters to Nature. 464 (7288): 587–91. PMID 20220755. doi:10.1038/nature08832.
- ↑ Bersaglieri, Todd; Sabeti, Pardis C.; Patterson, Nick; Vanderploeg, Trisha; Schaffner, Steve F.; Drake, Jared A.; Rhodes, Matthew; Reich, David E.; Hirschhorn, Joel N. (2004-06-01). "Genetic signatures of strong recent positive selection at the lactase gene". American Journal of Human Genetics. 74 (6): 1111–1120. ISSN 0002-9297. PMC 1182075 . PMID 15114531. doi:10.1086/421051.
- ↑ Tishkoff, Sarah A.; Reed, Floyd A.; Ranciaro, Alessia; Voight, Benjamin F.; Babbitt, Courtney C.; Silverman, Jesse S.; Powell, Kweli; Mortensen, Holly M.; Hirbo, Jibril B. (2007-01-01). "Convergent adaptation of human lactase persistence in Africa and Europe". Nature Genetics. 39 (1): 31–40. ISSN 1061-4036. PMC 2672153 . PMID 17159977. doi:10.1038/ng1946.
- ↑ Yi, Xin; Liang, Yu; Huerta-Sanchez, Emilia; Jin, Xin; Cuo, Zha Xi Ping; Pool, John E.; Xu, Xun; Jiang, Hui; Vinckenbosch, Nicolas (2010-07-02). "Sequencing of 50 human exomes reveals adaptation to high altitude". Science (New York, N.Y.). 329 (5987): 75–78. ISSN 1095-9203. PMC 3711608 . PMID 20595611. doi:10.1126/science.1190371.