Sample preparation in mass spectrometry
Sample preparation for mass spectrometry is used for the optimization of a sample for analysis in a mass spectrometer (MS). Each ionization method has certain factors that must be considered for that method to be successful, such as volume, concentration, sample phase, and composition of the analyte solution. Quite possibly the most important consideration in sample preparation is knowing what phase the sample must be in for analysis to be successful. In some cases the analyte itself must be purified before entering the ion source. In other situations, the matrix, or everything in the solution surrounding the analyte, is the most important factor to consider and adjust. Often, sample preparation itself for mass spectrometry can be avoided by coupling mass spectrometry to a chromatography method, or some other form of separation before entering the mass spectrometer. In some cases, the analyte itself must be adjusted so that analysis is possible, such as in protein mass spectrometry, where usually the protein of interest is cleaved into peptides before analysis, either by in-gel digestion or by proteolysis in solution.
Sample phase
| Sample Phase | Ionization method |
|---|---|
| Solid | Field desorption |
| Plasma desorption | |
| Fast Atom Bombardment (FAB) | |
| Secondary ion (SIMS) | |
| Solution | Matrix-assisted laser desorption ionization (MALDI) |
| Electrospray (ESI) | |
| Atmospheric pressure chemical ionization (APCI) | |
| Gas | Electron ionization |
| Photoionization | |
| Chemical ionization |
The first and most important step in sample preparation for mass spectrometry is determining what phase the sample needs to be in. Different ionization methods require different sample phases. Solid phase samples can be ionized through methods such as field desorption, plasma-desorption, fast atom bombardment, and secondary-ion ionization. Liquids with the analyte dissolved in them, or solutions, can be ionized through methods such as matrix-assisted laser desorption , electrospray ionization, and atmospheric-pressure chemical ionization.
Gas samples, or volatile samples, can be ionized using methods such as electron ionization, photoionization, and chemical ionization.
These lists are the most commonly used state of matter for each ionization method, but the ionization methods are not necessarily limited to these states of matter. For example, fast atom bombardment ionization is typically used to ionize solid samples, but this method is typically used on solids dissolved into solutions, and can also be used to analyze components that have entered the gas phase.[1][2][3]
Chromatography as a sample preparation method
In many mass spectrometry ionization methods, the sample must be in the liquid or gas phase for the ionization method to work. Sample preparation to ensure proper ionization can be difficult, but can be made easier by coupling the mass spectrometer to some chromatographic equipment. Gas chromatography(GC) or liquid chromatography(LC) can be used as a sample preparation method.
Gas chromatography

GC is a method involving the separation of different analytes within a sample of mixed gases. The separated gases can be detected multiple ways, but one of the most powerful detection methods for gas chromatography is mass spectrometry. After the gases separate, they enter the mass spectrometer and are analyzed. This combination not only separates the analytes, but gives structural information about each one. The GC sample must be volatile, or able to enter the gas phase, while also being thermally stable so that it does not break down as it is heated to enter the gas phase.[4][5] Mass spectrometry ionization techniques requiring the sample to be in the gas phase have similar concerns.
Electron ionization (EI) in mass spectrometry requires samples that are small molecules, volatile, and thermally stable, similar to that of gas chromatography. This ensures that as long as GC is performed on the sample before entering the mass spectrometer, the sample will be prepared for ionization by EI.[6][7]
Chemical ionization (CI) is another method that requires samples to be in the gas phase. This is so that the sample can react with a reagent gas to form an ion that can be analyzed by the mass spectrometer. CI has many of the same requirements in sample preparation as EI, such as volatility and thermal stability of the sample. GC is useful for sample preparation for this technique as well.[8] One advantage of CI is that larger molecules separated by GC can be analyzed by this ionization method. CI has a larger mass range than that of EI and can analyze molecules that EI may not be able to . CI also has the advantage of being less damaging to the sample molecule, so that less fragmentation occurs and more information about the original analyte can be determined.[6][9]
Photoionization (PI) was a method that was first applied as an ionization method to detecting gases separated by GC.[10] Years later, it was also applied as a detector for LC, though the samples must be vaporized first to be detected by the photoionization detector. Eventually PI was applied to mass spectrometry, particularly as an ionization method for gas chromatography-mass spectrometry.[11] Sample preparation for PI includes first ensuring the sample is in the gas phase. PI ionizes molecules by exciting the sample molecules with photons of light. This method only works if the sample and other components in the gas phase are excited by different wavelengths of light. It is important when preparing the sample, or photon source, that the wavelengths of ionization are adjusted to excite the sample analyte and nothing else.[6]
Liquid chromatography

Liquid chromatography (LC) is a method that in some ways is more powerful than GC, but can be coupled to mass spectrometry just as easily. In LC, the concerns involving sample preparation can be minimal. In LC, both the stationary and mobile phase can affect the separation, whereas in GC only the stationary phase should be influential. This allows for the sample preparation to be minimal if one is willing to adjust the stationary phase or mobile phase before running the sample. The primary concern is the concentration of analyte. If the concentration is too high then separation can be unsuccessful, but mass spectrometry as a detection method does not need complete separation, showing another benefit of coupling LC to a mass spectrometer.[12]
LC can be coupled to mass spectrometry through the vaporization of the liquid samples as they enter the mass spectrometer. This method can allow for ionization methods that require gaseous samples to be used, such as CI or PI, particularly atmospheric-pressure chemical ionization or atmospheric pressure photoionization, which allows for more interactions and more ionization.[6][13] Other ionization methods may not require the liquid sample to be vaporized, and can analyze the liquid sample itself. One example is fast-atom bombardment ionization which can allow for liquid samples separated by the LC to flow into the ionization chamber and be ionized easily.[1] The most common ionization method coupled to LC is some form of spray ionization, which includes thermospray ionization and more commonly, electrospray (ESI) ionization.
Thermospray was first developed as a way to effectively remove solvent and vaporize samples more easily. This method involves the liquid sample from the LC flowing through an electrically heated vaporizer that simply heats the sample, removing any solvent and therefore putting the sample in the gas phase.[14] Electrospray ionization (ESI) is similar to thermospray in the principle of removing the liquid solvent from the sample as much as possible, creating charged sample molecules either in small droplets or in gas form. Studies have shown that ESI can be as much as ten times more sensitive than other ionization methods coupled to LC.[12] The spray methods are particularly useful considering that non-volatile samples can be analyzed easily through this method since the sample is not itself turned into a gas, the liquid is simply removed, pushing the sample into a gaseous or mist phase.[14]
One sample preparation issue with liquid chromatography-mass spectrometry is possible matrix effects due to the presence of background molecules. These matrix effects have been shown to decrease the signal in methods such as PI and ESI by amounts as much as 60% depending on the sample being analyzed. The matrix effect can also cause an increase in signal, producing false positive results. This can be corrected by purifying the sample as much as possible before LC is performed, but in the case of analyzing environmental samples where everything in the sample is of concern, sample preparation may not be the ideal solution to fix the problem. Another method that can be applied to correct the issue is by using the standard addition method.[12][15]
Fast atom bombardment
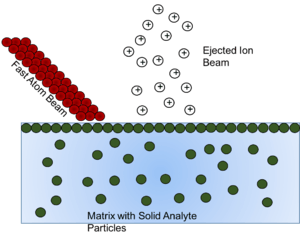
Fast atom bombardment (FAB) is a method involving using a beam of high energy atoms to strike a surface and generate ions. These solid analyte particles must be dissolved into some form of matrix, or non-volatile liquid to protect and assist in the ionization of the solid analyte. It has been shown that as the matrix is depleted, the ion formation diminishes, so choosing the right matrix compound is vital.[16]
The overall goal of the matrix compound is to present the sample to the atom beam at a high mobile surface concentration. For maximum sensitivity, the sample should form a perfect monolayer at the surface of a substrate having low volatility.[17] This monolayer effect can be seen in that once a certain concentration of analyte in matrix is reached, any concentration above that is seen to exhibit no effect, because once the monlayer is formed, any additional analyte is beneath the monolayer, and thus not affected by the atom beam. The concentration needed to cause this effect is seen to change as the amount of non-volatile matrix changes. So concentration of solid analyte needs to be considered in the preparation of the solution for analysis so that signal from "hidden" analyte is not missed.[17]
To choose the matrix for each solid analyte, three criteria must be considered. First, it should dissolve the solid compound to be analysed (with or without the aid of a cosolvent or additive), thus allowing molecules of that compound to diffuse to the surface layers, replenishing the sample molecules that have been ionized or destroyed by interaction with the fast atom beam. It should also be noted that another mechanism for explanation of ion formation in FAB involves the idea that sputtering occurs from the bulk rather than the surface, but in that case, the solubility is still largely important to insure homogeneity of solid analyte in the bulk solution.[16][17] Secondly, the matrix should have a low volatility under the conditions of the mass spectrometer. As mentioned above, as the matrix is depleted, the ionization decreases as well, so maintaining the matrix is vital. Thirdly, the matrix should not react with the solid analyte in question, or if it does react, it should be in an understood and reproducible way.[16] This ensures reproducibility of analysis and identification of the actual analyte rather than a derivative of the analyte.
The most commonly used compounds as a matrix are variations of glycerol, such as glycerol, deuteroglycerol, thioglycerol, and aminoglycerol. If the sample cannot dissolve in the chosen matrix, such as glycerol, a cosolvent or additive can be mixed with the matrix to facilitate the dissolving of the solid analyte. For example, chlorophyll A is completely insoluble in glycerol, but by mixing in a small amount of Triton X-100, a derivative of polyethylene glycol, the chlorophyll becomes highly soluble within the matrix.[17][18] It is important to note that though a good signal may be achieved through glycerol or glycerol with an additive, there could be other matrix compounds that can offer an even better signal. Optimization of matrix compounds and concentration of solid analyte are vital for FAB measurements.
Secondary ion mass spectrometry
Secondary ion mass spectrometry (SIMS) is a method very similar to FAB in that a beam of particles is fired against the surface of a sample in order to cause sputtering, in which the molecules of the sample ionize and leave the surface, thus allowing for the ions or the sample to be analyzed. The primary difference is that in SIMS, an ion beam is fired against the surface, but in FAB, an atom beam is fired against the surface. The other primary difference, of more interest to this page, is that, unlike FAB, SIMS is typically performed on a solid sample with little sample preparation required.[19]
The main consideration with SIMS is ensuring that the sample is stable under ultra-high vacuum, or pressures less than 10−8 torr. The nature of the ultra-high vacuum is that it ensures the sample remains constant during analysis as well as ensuring the high energy ion beam strikes the sample. Ultra-high vacuum solves many of the problems that need to be considered during sample preparation.[20] When preparing the sample for analysis, another thing that should be considered is the thickness of the film. Typically, if a thin monolayer can be deposited onto the surface of a noble metal, analysis should be successful.[21] If the film thickness is too large, which is common in real world analysis, the problem can be solved by methods such as depositing a perforated silver foil over a nickel grid onto the film surface. This yields similar results to thin films deposited directly onto a noble metal.[19]
Matrix-assisted laser desorption/ionization
For matrix-assisted laser desorption/ionization (MALDI) mass spectrometry a solid or liquid sample is mixed with a matrix solution, to help the sample avoid processes such as aggregation or precipitation, while helping the sample remain stable during the ionization process.[22][23] The matrix crystallizes with the sample and is then deposited on a sample plate, which can be made of a range of materials, from inert metals to inert polymers. The matrix containing the sample molecules is then transferred to the gas phase by pulsed laser irradiation. The makeup of the matrix, interactions between the sample and the matrix, and how the sample is deposited are all extremely important during sample preparation to ensure the best possible results.
The selection of a matrix is the first step when preparing samples for MALDI analysis. The primary goals of the matrix are to absorb the energy from a laser, thus transferring it to the analyte molecules, and to separate the analyte molecules from each other.[24] A consideration that should be taken into account when choosing a matrix is what type of analyte ion is expected or desired. Knowing the acidity or basicity of the analyte molecule compared with the acidity or basicity of the matrix, for example, is valuable knowledge when choosing a matrix. The matrix should not compete with the analyte molecule, so the matrix should not want to form the same type of ion as the analyte. For example, if the desired analyte has a high amount of acidity, it would be logical to choose a matrix with a high amount of basicity to avoid competition and facilitate the formation of an ion.[25] The pH of the matrix can also be used to select what sample you want to obtain spectra for. For example, in the case of proteins, a very acidic pH can show very little of the peptide components, but can show very good signal for those components that are larger. If the pH is increased towards a more basic pH, then smaller components become easier to see.[26]
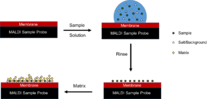
The concentration of salt in the sample is a factor that needs to be considered when preparing a MALDI sample as well. Salts can aid a MALDI spectra by preventing aggregation or precipitation while stabilizing the sample. However, interfering signals can be observed due to side reactions of the matrix with the sample, such as in the case of the matrix interacting with alkali metal ions which can impair the analysis of the spectra. Typically the amount of salt in the matrix only becomes a problem in very high concentrations, such as 1 molar.[23] The problem of having too high a concentration of salt in the sample can be solved by first running the solution through liquid chromatography to help purify the sample, but this method is time consuming and results in the loss of some of the sample to be analyzed. Another method is focused on purification once the sample solution is deposited onto the sample probe. Many sample probes can be designed to have a membrane on the surface that can selectively bind the sample in question to the probe surface. The surface can then be rinsed off to remove all unnecessary salts or background molecules. The matrix of appropriate salt concentration can then be deposited directly onto the sample on the probe surface and crystallized there.[23] Despite these negative effects of salt concentration, a separate desalting step is usually not necessary in the case of proteins, because the selection of appropriate buffer salts prevents the occurrence of this problem.[27]
How the sample and matrix is deposited on the surface of the sample probe needs to be a consideration in sample preparation as well. The dried drop method is the simplest of deposition methods. The matrix and sample solution are mixed together and then a small drop of the mixture is placed on the sample probe surface and allowed to dry, thus crystallizing. The sandwich method involves depositing a layer of matrix onto the surface of the probe and allowing it to dry. A drop of the sample followed by a drop of additional matrix is then applied to the layer of dried matrix and allowed to dry as well.[28] Variations on the sandwich technique involve depositing the matrix on the surface and then depositing the sample directly on top of the matrix. A particularly useful method involves depositing the matrix solution on the surface of the sample probe in a solvent that will evaporate very rapidly, thus forming a very thin fine layer of matrix. The sample solution is then placed on top of the matrix layer and allowed to evaporate slowly, thus integrating the sample into the top layer of matrix as the sample solution evaporates.[29] An addition concern when depositing the sample on the surface of the probe is the solubility of the sample in the matrix. If the sample is insoluble in the matrix, additional methods must be employed. A method used in this case involves mechanical grinding and mixing of solid sample and solid matrix crystals. Once blended well, this powder can be deposited on the surface of the sample probe in free powder form or as a pill. Another possible method is placing the sample on the surface of the probe and applying vaporized matrix to the sample probe to allow the matrix to condense around the sample.[30]
Electrospray ionization
Electrospray ionization (ESI) is a technique that involves using high voltages to create an electrospray, or a fine aerosol created by the high voltages.[31] ESI sample preparation can be very important and the quality of results can be heavily determined by the characteristics of the sample.[32] ESI experiments can be run on-line or off-line. In on-line measurements the mass spectrometer is connected to a liquid chromatograph and as the samples are separated they are ionized into the mass spectrometer by the ESI system; sample preparation is actually performed before the LC separation.[33] In off-line measurements, the analyte solution is applied directly to the mass spectrometer by a spray capillary . Off-line sample preparation has many considerations, such as the fact that the capillary used allows for the application of volumes in the nanoliter range, which can contain a concentration too small for analysis of many compounds, such as proteins. An additional problem can be loss of ESI signal due to interference between the analyte sample and background components. Unfortunately, it has been shown that sample preparation itself can only slightly alleviate this problem which is due more to the nature of the analyte itself than the preparation.[34] In ESI the principle problem comes not from reactions in the gas phase but rather from problems involving the solution phase of the droplets themselves. Issues can be due to non-volatile substances remaining in the drops, which can change the efficiency of droplet formation or droplet evaporation, which in turn affects the amount of charged ions in the gas phase that ultimately reach the mass spectrometer. These problems can be fixed in multiple ways, including increasing the amount of concentration of analyte compared to matrix in the sample solution or by running the sample through a more extensive chromatographic technique before analysis.[35][36] An example of a chromatographic technique that can aid in signal in ESI involves using 2-D liquid chromatography, or running the sample through two separate chromatography columns, giving better separation of the analyte from the matrix.[37][38]

ESI variations

There are some ESI methods that require little to no sample preparation. One such method is a method termed extractive electrospray ionization (EESI). This method involves having an electrospray of solvent directed at an angle against a different spray of the sample solution, produced by a separate nebulizer. This method requires no sample preparation in that the electrospray of solvent extracts the sample from the complex mixture, effectively removing any background contaminants.[39] Another particularly powerful variation on ESI is desorption electrospray ionization (DESI), which involves directing an electrospray at a surface with the sample deposited on top of it. The sample is ionized in the electrospray as it splashes off the surface, then traveling to the mass spectrometer. This method is important because of the fact that no sample preparation is needed for this method. A sample simply needs to be deposited on a surface, such as paper.[40] Atmospheric pressure chemical ionization (APCI) is similar to ESI in that the sample is nebulized in droplets that are then evaporated, leaving behind a charged ion to be analyzed. APCI experiences few of the negative matrix effects experienced by ESI due to the fact that ionization occurs in the gas phase in this method rather than the within the liquid droplets as in ESI and the fact that in APCI there is an overabundance of reaction gas, thus minimizing the effect of the matrix on the ionization process.[41][42]
Protein ESI
A major application for ESI is the field of protein mass spectrometry. Here, the MS is used for the identification and sizing of proteins. The identification of a protein sample can be done in an ESI-MS by de novo peptide sequencing (using tandem mass spectrometry) or peptide mass fingerprinting. Both methods require the previous digestion of proteins to peptides, mostly accomplished enzymatically using proteases. As well for the digestion in solution as for the in-gel digestion buffered solutions are needed, whose content in salts is too high and in analyte is too low for a successful ESI-MS measurement. Therefore, a combined desalting and concentration step is performed. Usually a reversed phase liquid chromatography is used, in which the peptides stay bound to the chromatography matrix whereas the salts are removed by washing. The peptides can be eluted from the matrix by the use of a small volume of a solution containing a large portion of organic solvent, which results in the reduction of the final volume of the analyte. In LC-MS the desalting/concentration is realised with a pre-column, in off-line measurements reversed phase micro columns are used, which can be used directly with microliter pipettes. Here, the peptides are eluted with the spray solution containing an appropriate portion of organic solvent. The resulting solution (usually a few microliters)is enriched with the analyte and, after transfer to the spray capillary, can be directly used in the MS.
See also
References
- 1 2 Caprioli, Richard M. (1990-04-15). "Continuous-Flow Fast Atom Bombardment Mass Spectrometry". Analytical Chemistry. 62 (8): 477A–485A. doi:10.1021/ac00207a715.
- ↑ Takayama, Mitsuo (1995-07-21). "Gas-phase fast-atom bombardment mass spectrometry". International Journal of Mass Spectrometry and Ion Processes. 152: 1–20. doi:10.1016/0168-1176(95)04298-9.
- ↑ Kralj, B.; Kramer, V.; Vrscaj, V. (1983). "Fast Atom Bombardment of Molecules in the Gaseous State". International Journal of Mass Spectrometry and Ion Physics. 46: 399–402. doi:10.1016/0020-7381(83)80136-3.
- ↑ James M. Miller (16 December 2005). Chromatography: Concepts and Contrasts. John Wiley & Sons. ISBN 978-0-471-98059-9.
- ↑ Mondello, Luigi; Tranchida, Peter Quinto; Dugo, Paola; Dugo, Giovanni (2008). "Comprehensive two-dimensional gas chromatography-mass spectrometry: A review". Mass Spectrometry Reviews. 27 (2): 101–124. ISSN 0277-7037. doi:10.1002/mas.20158.
- 1 2 3 4 Chhabil Dass (11 May 2007). Fundamentals of Contemporary Mass Spectrometry. John Wiley & Sons. ISBN 978-0-470-11848-1.
- ↑ Aiken, Allison C.; DeCarlo, Peter F.; Jimenez, Jose L. (2007). "Elemental Analysis of Organic Species with Electron Ionization High-Resolution Mass Spectrometry". Analytical Chemistry. 79 (21): 8350–8358. ISSN 0003-2700. PMID 17914892. doi:10.1021/ac071150w.
- ↑ Angerosa, Franca; d'Alessandro, Nicola; Corana, Federica; Mellerio, Giorgio (1996). "Characterization of phenolic and secoiridoid aglycons present in virgin olive oil by gas chromatography-chemical ionization mass spectrometry". Journal of Chromatography A. 736 (1–2): 195–203. ISSN 0021-9673. doi:10.1016/0021-9673(95)01375-X.
- ↑ Field, Frank H. (1968). "Chemical ionization mass spectrometry". Accounts of Chemical Research. 1 (2): 42–49. ISSN 0001-4842. doi:10.1021/ar50002a002.
- ↑ Driscoll, John N. (1977). "Evaluation of a new photoionization detector for organic compounds". Journal of Chromatography A. 134 (1): 49–55. ISSN 0021-9673. doi:10.1016/S0021-9673(00)82568-6.
- ↑ Raffaelli, Andrea; Saba, Alessandro (2003). "Atmospheric pressure photoionization mass spectrometry". Mass Spectrometry Reviews. 22 (5): 318–331. ISSN 0277-7037. doi:10.1002/mas.10060.
- 1 2 3 Petrović, Mira; Hernando, Maria Dolores; Díaz-Cruz, M. Silvia; Barceló, Damià (2005). "Liquid chromatography–tandem mass spectrometry for the analysis of pharmaceutical residues in environmental samples: a review". Journal of Chromatography A. 1067 (1–2): 1–14. ISSN 0021-9673. doi:10.1016/j.chroma.2004.10.110.
- ↑ Robb, Damon B.; Covey, Thomas R.; Bruins, Andries P. (2000). "Atmospheric Pressure Photoionization: An Ionization Method for Liquid Chromatography−Mass Spectrometry". Analytical Chemistry. 72 (15): 3653–3659. ISSN 0003-2700. doi:10.1021/ac0001636.
- 1 2 Blakley, C. R.; Vestal, M. L. (1983). "Thermospray interface for liquid chromatography/mass spectrometry". Analytical Chemistry. 55 (4): 750–754. ISSN 0003-2700. doi:10.1021/ac00255a036.
- ↑ Benijts, Tom; Dams, Riet; Lambert, Willy; De Leenheer, André (2004). "Countering matrix effects in environmental liquid chromatography–electrospray ionization tandem mass spectrometry water analysis for endocrine disrupting chemicals". Journal of Chromatography A. 1029 (1–2): 153–159. ISSN 0021-9673. doi:10.1016/j.chroma.2003.12.022.
- 1 2 3 Gower, John Leveson (1985). "Matrix compounds for fast atom bombardment mass spectrometry". Biological Mass Spectrometry. 12 (5): 191–196. ISSN 1052-9306. doi:10.1002/bms.1200120502.
- 1 2 3 4 Barber, Michael; Bordoli, Robert S.; Elliott, Gerald J.; Sedgwick, R. Donald; Tyler, Andrew N. (1982). "Fast Atom Bombardment Mass Spectrometry". Analytical Chemistry. 54 (4): 645A–657A. doi:10.1021/ac00241a817.
- ↑ product page from Shun Chia
- 1 2 Van Vaeck, Luc; Adriaens, Annemie; Gijbels, Renaat (1999-04-28). "Static Secondary Ion Mass Spectrometry: (S-SIMS) Part 1. Methodology and Structural Interpretation". Mass Spectrometry Reviews. 18: 1–47. doi:10.1002/(sici)1098-2787(1999)18:1<1::aid-mas1>3.3.co;2-n.
- ↑ Belu, Anna M.; Graham, Daniel J.; Castner, David G. (2003). "Time-of-flight secondary ion mass spectrometry: techniques and applications for the characterization of biomaterial surfaces". Biomaterials. 24 (21): 3635–3653. ISSN 0142-9612. doi:10.1016/S0142-9612(03)00159-5.
- ↑ Benninghoven, Alfred; Hagenhoff, Birgit; Niehuis, Ewald (1993-07-15). "Surface MS: Probing Real World Samples". Analytical Chemistry. 65: 630A–640A. doi:10.1021/ac00062a002.
- ↑ Fenyo D; Wang Q; DeGrasse JA; Padovan JC; Cadene M; Chait BT (2007). "MALDI sample preparation: the ultra thin layer method". J Vis Exp (3): 192. PMC 2535834
 . PMID 18978997. doi:10.3791/192.
. PMID 18978997. doi:10.3791/192. - 1 2 3 Xu, Yingda; Bruening, Merlin L.; Watson, J. Throck (2003). "Non-specific, on-probe cleanup methods for MALDI-MS samples". Mass Spectrometry Reviews. 22 (6): 429–440. ISSN 0277-7037. PMID 14528495. doi:10.1002/mas.10064.
- ↑ Hillenkamp, Franz; Karas, Michael; Beavis, Ronald C.; Chait, Brian T. (1991). "Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry of Biopolymers". Analytical Chemistry. 63 (24): 1193A–1203A. ISSN 0003-2700. PMID 1789447. doi:10.1021/ac00024a716.
- ↑ Zenobi, Renato; Knochenmuss, Richard (1998). "Ion formation in MALDI mass spectrometry". Mass Spectrometry Reviews. 17 (5): 337–366. ISSN 0277-7037. doi:10.1002/(SICI)1098-2787(1998)17:5<337::AID-MAS2>3.0.CO;2-S.
- ↑ Cohen, Steven L.; Chait, Brian T. (1996). "Influence of Matrix Solution Conditions on the MALDI-MS Analysis of Peptides and Proteins". Analytical Chemistry. 68 (1): 31–37. ISSN 0003-2700. doi:10.1021/ac9507956.
- ↑ Smirnov et al., Anal. Chem., 76 (10), S. 2958-2965, 2004
- ↑ Kussmann, Martin; Nordhoff, Eckhard; Rahbek-Nielsen, Henrik; Haebel, Sophie; Rossel-Larsen, Martin; Jakobsen, Lene; Gobom, Johan; Mirgorodskaya, Ekatarina; Kroll-Kristensen, Anne; Palm‖, Lisbeth; Roepstorff, Peter (1997). "Matrix-assisted Laser Desorption/Ionization Mass Spectrometry Sample Preparation Techniques Designed for Various Peptide and Protein Analytes". Journal of Mass Spectrometry. 32 (6): 593–601. ISSN 1076-5174. doi:10.1002/(SICI)1096-9888(199706)32:6<593::AID-JMS511>3.0.CO;2-D.
- ↑ Vorm, Ole.; Roepstorff, Peter.; Mann, Matthias. (1994). "Improved Resolution and Very High Sensitivity in MALDI TOF of Matrix Surfaces Made by Fast Evaporation". Analytical Chemistry. 66 (19): 3281–3287. ISSN 0003-2700. doi:10.1021/ac00091a044.
- ↑ Trimpin, S.; Keune, S.; Räder, H. J.; Müllen, K. (2006). "Solvent-free MALDI-MS: Developmental improvements in the reliability and the potential of MALDI in the analysis of synthetic polymers and giant organic molecules". Journal of the American Society for Mass Spectrometry. 17 (5): 661–671. ISSN 1044-0305. doi:10.1016/j.jasms.2006.01.007.
- ↑ Ho, CS; Chan MHM; Cheung RCK; Law LK; Lit LCW; Ng KF; Suen MWM; Tai HL (February 2003). "Electrospray Ionisation Mass Spectrometry: Principles and Clinical Applications". Clin Biochem Rev. 24 (1): 3–12. PMC 1853331 . PMID 18568044.
- ↑ Du L; White RL (November 2008). "Improved partition equilibrium model for predicting analyte response in electrospray ionization mass spectrometry". J Mass Spectrom. 44 (2): 222–9. PMID 19003789. doi:10.1002/jms.1501.
- ↑ Pitt, James J (February 2009). "Principles and Applications of Liquid Chromatography-Mass Spectrometry in Clinical Biochemistry". Clin Biochem Rev. 30 (1): 19–34. PMC 2643089 . PMID 19224008.
- ↑ Bonfiglio, Ryan; King, Richard C.; Olah, Timothy V.; Merkle, Kara (1999). "The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds". Rapid Communications in Mass Spectrometry. 13 (12): 1175–1185. ISSN 0951-4198. doi:10.1002/(SICI)1097-0231(19990630)13:12<1175::AID-RCM639>3.0.CO;2-0.
- ↑ King, Richard; Bonfiglio, Ryan; Fernandez-Metzler, Carmen; Miller-Stein, Cynthia; Olah, Timothy (2000). "Mechanistic investigation of ionization suppression in electrospray ionization". Journal of the American Society for Mass Spectrometry. 11 (11): 942–950. ISSN 1044-0305. doi:10.1016/S1044-0305(00)00163-X.
- ↑ Annesley, T. M. (2003). "Ion Suppression in Mass Spectrometry". Clinical Chemistry. 49 (7): 1041–1044. ISSN 0009-9147. PMID 12816898. doi:10.1373/49.7.1041.
- ↑ Pascoe, Rob; Foley, Joe P.; Gusev, Arkady I. (2001). "Reduction in Matrix-Related Signal Suppression Effects in Electrospray Ionization Mass Spectrometry Using On-Line Two-Dimensional Liquid Chromatography". Analytical Chemistry. 73 (24): 6014–6023. ISSN 0003-2700. doi:10.1021/ac0106694.
- ↑ Taylor, Paul J. (2005). "Matrix effects: the Achilles heel of quantitative high-performance liquid chromatography–electrospray–tandem mass spectrometry". Clinical Biochemistry. 38 (4): 328–334. ISSN 0009-9120. doi:10.1016/j.clinbiochem.2004.11.007.
- ↑ Chen, Huanwen; Venter, Andre; Cooks, R. Graham (2006). "Extractive electrospray ionization for direct analysis of undiluted urine, milk and other complex mixtures without sample preparation". Chemical Communications (19): 2042. ISSN 1359-7345. doi:10.1039/b602614a.
- ↑ Chen, Huanwen; Pan, Zhengzheng; Talaty, Nari; Raftery, Daniel; Cooks, R. Graham (2006). "Combining desorption electrospray ionization mass spectrometry and nuclear magnetic resonance for differential metabolomics without sample preparation". Rapid Communications in Mass Spectrometry. 20 (10): 1577–1584. ISSN 0951-4198. doi:10.1002/rcm.2474.
- ↑ Souverain, Sandrine; Rudaz, Serge; Veuthey, Jean-Luc (2004). "Matrix effect in LC-ESI-MS and LC-APCI-MS with off-line and on-line extraction procedures". Journal of Chromatography A. 1058 (1–2): 61–66. ISSN 0021-9673. doi:10.1016/S0021-9673(04)01477-3.
- ↑ Matuszewski, B. K.; Constanzer, M. L.; Chavez-Eng, C. M. (2003). "Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS". Analytical Chemistry. 75 (13): 3019–3030. ISSN 0003-2700. doi:10.1021/ac020361s.