RNA-Seq
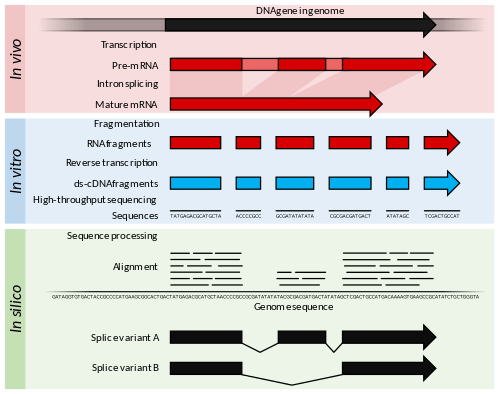
RNA-Seq (RNA sequencing), also called whole transcriptome shotgun sequencing[1] (WTSS), uses next-generation sequencing (NGS) to reveal the presence and quantity of RNA in a biological sample at a given moment in time.[2][3]
RNA-Seq is used to analyze the continually changing cellular transcriptome. Specifically, RNA-Seq facilitates the ability to look at alternative gene spliced transcripts, post-transcriptional modifications, gene fusion, mutations/SNPs and changes in gene expression over time, or differences in gene expression in different groups or treatments.[4] In addition to mRNA transcripts, RNA-Seq can look at different populations of RNA to include total RNA, small RNA, such as miRNA, tRNA, and ribosomal profiling.[5] RNA-Seq can also be used to determine exon/intron boundaries and verify or amend previously annotated 5' and 3' gene boundaries.
Prior to RNA-Seq, gene expression studies were done with hybridization-based microarrays. Issues with microarrays include cross-hybridization artifacts, poor quantification of lowly and highly expressed genes, and needing to know the sequence a priori.[6] Because of these technical issues, transcriptomics transitioned to sequencing-based methods. These progressed from Sanger sequencing of Expressed Sequence Tag libraries, to chemical tag-based methods (e.g., serial analysis of gene expression), and finally to the current technology, next-gen sequencing of cDNA (notably RNA-Seq).
Methods
Library preparation

The general steps to prepare a complementary DNA (cDNA) library for sequencing are described below, but often vary between platforms.[7][8][9]
- RNA Isolation: RNA is isolated from tissue and mixed with deoxyribonuclease (DNase). DNase reduces the amount of genomic DNA. The amount of RNA degradation is checked with gel and capillary electrophoresis and is used to assign an RNA integrity number to the sample. This RNA quality and the total amount of starting RNA are taken into consideration during the subsequent library preparation, sequencing, and analysis steps.
- RNA selection/depletion: To analyze signals of interest, the isolated RNA can either be kept as is, filtered for RNA with 3' polyadenylated (poly(A)) tails to include only mRNA, depleted of ribosomal RNA (rRNA), and/or filtered for RNA that binds specific sequences (Table). The RNA with 3' poly(A) tails are mature, processed, coding sequences. Poly(A) selection is performed by mixing RNA with poly(T) oligomers covalently attached to a substrate, typically magnetic beads.[1][10][11] Poly(A) selection ignores noncoding RNA and introduces 3' bias,[12] which is avoided with the ribosomal depletion strategy. The rRNA is removed because it represents over 90% of the RNA in a cell, which if kept would drown out other data in the transcriptome.
- cDNA synthesis: DNA sequencing technology is more mature, so the RNA is reverse transcribed to cDNA. Reverse transcription results in loss of strandedness, which can be avoided with chemical labelling. Fragmentation and size selection are performed to purify sequences that are the appropriate length for the sequencing machine. The RNA, cDNA, or both are fragmented with enzymes, sonication, or nebulizers. Fragmentation of the RNA reduces 5' bias of randomly primed-reverse transcription and the influence of primer binding sites,[10] with the downside that the 5' and 3' ends are converted to DNA less efficiently. Fragmentation is followed by size selection, where either small sequences are removed or a tight range of sequence lengths are selected. Because small RNAs like miRNAs are lost, these are analyzed independently. The cDNA for each experiment can be indexed with a hexamer or octamer barcode, so that these experiments can be pooled into a single lane for multiplexed sequencing.
| Strategy | Type of RNA | Ribosomal RNA content | Unprocessed RNA content | Genomic DNA content | Isolation method |
|---|---|---|---|---|---|
| Total RNA | All | High | High | High | None |
| PolyA selection | Coding | Low | Low | Low | Hybridization with poly(dT) oligomers |
| rRNA depletion | Coding, noncoding | Low | High | High | Removal of oligomers complementary to rRNA |
| RNA capture | Targeted | Low | Moderate | Low | Hybridization with probes complementary to desired transcripts |
Small RNA/non-coding RNA sequencing
When sequencing RNA other than mRNA, the library preparation is modified. The cellular RNA is selected based on the desired size range. For small RNA targets, such as miRNA, the RNA is isolated through size selection. This can be performed with a size exclusion gel, through size selection magnetic beads, or with a commercially developed kit. Once isolated, linkers are added to the 3' and 5' end then purified. The final step is cDNA generation through reverse transcription.
Direct RNA sequencing


As converting RNA into cDNA using reverse transcriptase has been shown to introduce biases and artifacts that may interfere with both the proper characterization and quantification of transcripts,[13] single molecule Direct RNA Sequencing (DRSTM) technology was under development by Helicos (now bankrupt). DRSTM sequences RNA molecules directly in a massively-parallel manner without RNA conversion to cDNA or other biasing sample manipulations such as ligation and amplification.
Experimental considerations
A variety of parameters are considered when designing and conducting RNA-Seq experiments:
- Tissue specificity: Gene expression varies within and between tissues, and RNA-Seq measures this mix of cell types. This may make it difficult to isolate the biological mechanism of interest. Single cell sequencing can be used to study each cell individually, mitigating this issue.
- Time dependence: Gene expression changes over time, and RNA-Seq only takes a snapshot. Time course experiments can be performed to observe changes in the transcriptome.
- Coverage (also known as depth): RNA harbors the same mutations observed in DNA, and detection requires deeper coverage. With high enough coverage, RNA-Seq can be used to estimate the expression of each allele. This may provide insight into phenomena such as imprinting or cis-regulatory effects. The depth of sequencing required for specific applications can be extrapolated from a pilot experiment.[14]
- Data generation artifacts (also known as technical variance): The reagents (e.g., library preparation kit), personnel involved, and type of sequencer (e.g., Illumina, Pacific Biosciences) can result in technical artifacts that might be mis-interpreted as meaningful results. As with any scientific experiment, it is prudent to conduct RNA-Seq in a well controlled setting. If this is not possible or the study is a meta-analysis, another solution is to detect technical artifacts by inferring latent variables (typically principal component analysis or factor analysis) and subsequently correcting for these variables.[15]
- Data management: A single RNA-Seq experiment in humans is usually on the order of 1 Gb.[16] This large volume of data can pose storage issues. One solution is compressing the data using multi-purpose computational schemas (e.g., gzip) or genomics-specific schemas. The latter can be based on reference sequences or de novo. Another solution is to perform microarray experiments, which may be sufficient for hypothesis-driven work or replication studies (as opposed to exploratory research).
Analysis

Transcriptome assembly
Two methods are used to assign raw sequence reads to genomic features (i.e., assemble the transcriptome):
- De novo: This approach does not require a reference genome to reconstruct the transcriptome, and is typically used if the genome is unknown, incomplete, or substantially altered compared to the reference.[17] Challenges when using short reads for de novo assembly include 1) determining which reads should be joined together into contiguous sequences (contigs), 2) robustness to sequencing errors and other artifacts, and 3) computational efficiency. The primary algorithm used for de novo assembly transitioned from overlap graphs, which identify all pair-wise overlaps between reads, to de Bruijn graphs, which break reads into sequences of length k and collapse all k-mers into a hash table.[18] Overlap graphs were used with Sanger sequencing, but do not scale well to the millions of reads generated with RNA-Seq. Examples of assemblers that use de Bruijn graphs are Velvet,[19] Trinity,[17] Oases,[20] and Bridger.[21] Paired end and long read sequencing of the same sample can mitigate the deficits in short read sequencing by serving as a template or skeleton. Metrics to assess the quality of a de novo assembly include median contig length, number of contigs and N50.[22]
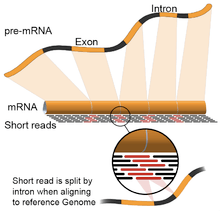
- Genome guided: This approach relies on the same methods used for DNA alignment, with the additional complexity of aligning reads that cover non-continuous portions of the reference genome.[23] These non-continuous reads are the result of sequencing spliced transcripts (see figure). Typically, alignment algorithms have two steps: 1) align short portions of the read (i.e., seed the genome), and 2) use dynamic programming to find an optimal alignment, sometimes in combination with known annotations. Software tools that use genome-guided alignment include Bowtie,[24] TopHat (which builds on BowTie results to align splice junctions),[25][26] Subread,[27] STAR,[23] Sailfish,[28] Kallisto,[29] and GMAP.[30] The quality of a genome guided assembly can be measured with both 1) de novo assembly metrics (e.g., N50) and 2) comparisons to known transcript, splice junction, genome, and protein sequences using precision, recall, or their combination (e.g., F1 score).[22]
A note on assembly quality: The current consensus is that 1) assembly quality can vary depending on which metric is used, 2) assemblies that scored well in one species do not necessarily perform well in the other species, and 3) combining different approaches might be the most reliable.[31][32]
Gene expression
Expression is quantified to study cellular changes in response to external stimuli, differences between healthy and diseased states, and other research questions. Gene expression is often used as a proxy for protein abundance, but these are often not equivalent due to post transcriptional events such as RNA interference and nonsense-mediated decay.[33]
Expression is quantified by counting the number of reads that mapped to each locus in the transcriptome assembly step. Expression can be quantified for exons or genes using contigs or reference transcript annotations.[7] These observed RNA-Seq read counts have been robustly validated against older technologies, including expression microarrays and qPCR.[14][34] Tools that quantify counts are HTSeq,[35] FeatureCounts,[36] Rcount,[37] maxcounts,[38] FIXSEQ,[39] and Cuffquant. The read counts are then converted into appropriate metrics for hypothesis testing, regressions, and other analyses. Parameters for this conversion are:
- Library size: Although sequencing depth is pre-specified when conducting multiple RNA-Seq experiments, it will still vary widely between experiments.[40] Therefore, the total number of reads generated in a single experiment (library size) is typically adjusted by converting counts to fragments, reads, or counts per million mapped reads (FPM, RPM, or CPM).
- Gene length: Longer genes will have more fragments/reads/counts than shorter genes if transcript expression is the same. This is adjusted by dividing the FPM by the length of a gene, resulting in the metric fragments per kilobase of transcript per million mapped reads (FPKM).[41]
- Total sample RNA output: Because the same amount of RNA is extracted from each sample, samples with more total RNA will have less RNA per gene. These genes appear to have decreased expression, resulting in false positives in downstream analyses.[42]
- Variance for each gene's expression: is modeled to account for sampling error (important for genes with low read counts), increase power, and decrease false positives. Variance can be estimated as a normal, Poisson, or negative binomial distribution.[43][44][45]
Differential expression and absolute quantification of transcripts
RNA-Seq is generally used to compare gene expression between conditions, such as a drug treatment vs non-treated, and find out which genes are up- or down-regulated in each condition. In principle, RNA-Seq will make it possible to account for all the transcripts in the cell for each condition. Differently expressed genes can be identified using tools that count the sequencing reads per gene and compare them between samples. Many packages are available for this type of analysis;[46] some of the most commonly used tools are DESeq[47] and edgeR,[48] packages from Bioconductor.[49][50] Both these tools use a model based on the negative binomial distribution.[47][48]
It is not possible to do absolute quantification using the common RNA-Seq pipeline, because it only provides RNA levels relative to all transcripts. If the total amount of RNA in the cell changes between conditions, relative normalization will misrepresent the changes for individual transcripts. Absolute quantification of mRNAs is possible by performing RNA-Seq with added spike ins, samples of RNA at known concentrations. After sequencing, the read count of the spike ins sequences is used to determine the direct correspondence between read count and biological fragments.[51][52] In developmental studies, this technique has been used in Xenopus tropicalis embryos at a high temporal resolution, to determine transcription kinetics.[53]
Coexpression networks
Coexpression networks are data-derived representations of genes behaving in a similar way across tissues and experimental conditions.[54] Their main purpose lies in hypothesis generation and guilt-by-association approaches for inferring functions of previously unknown genes.[54] RNASeq data has been recently used to infer genes involved in specific pathways based on Pearson correlation, both in plants [55] and mammals.[56] The main advantage of RNASeq data in this kind of analysis over the microarray platforms is the capability to cover the entire transcriptome, therefore allowing the possibility to unravel more complete representations of the gene regulatory networks. Differential regulation of the splice isoforms of the same gene can be detected and used to predict and their biological functions.[57][58] Weighted gene co-expression network analysis has been successfully used to identify co-expression modules and intramodular hub genes based on RNA seq data. Co-expression modules may corresponds to cell types or pathways. Highly connected intramodular hubs can be interpreted as representatives of their respective module. Variance-Stabilizing Transformation approaches for estimating correlation coefficients based on RNA seq data have been proposed.[55]
Single nucleotide variation discovery
Transcriptome single nucleotide variation has been analyzed in maize on the Roche 454 sequencing platform.[59] Directly from the transcriptome analysis, around 7000 single nucleotide polymorphisms (SNPs) were recognized. Following Sanger sequence validation, the researchers were able to conservatively obtain almost 5000 valid SNPs covering more than 2400 maize genes. RNA-seq is limited to transcribed regions however, since it will only discover sequence variations in exon regions. This misses many subtle but important intron alleles that affect disease such as transcription regulators, leaving analysis to only large effectors. While some correlation exists between exon to intron variation, only whole genome sequencing would be able to capture the source of all relevant SNPs.[60]
The only way to be absolutely sure of the individual's mutations is to compare the transcriptome sequences to the germline DNA sequence. This enables the distinction of homozygous genes versus skewed expression of one of the alleles and it can also provide information about genes that were not expressed in the transcriptomic experiment. An R-based statistical package known as CummeRbund[61] can be used to generate expression comparison charts for visual analysis.
Post-transcriptional SNVs
Having the matching genomic and transcriptomic sequences of an individual can also help in detecting post-transcriptional edits,[8] where, if the individual is homozygous for a gene, but the gene's transcript has a different allele, then a post-transcriptional modification event is determined.
mRNA centric single nucleotide variants (SNVs) are generally not considered as a representative source of functional variation in cells, mainly due to the fact that these mutations disappear with the mRNA molecule, however the fact that efficient DNA correction mechanisms do not apply to RNA molecules can cause them to appear more often. This has been proposed as the source of certain prion diseases,[62] also known as TSE or transmissible spongiform encephalopathies.

Fusion gene detection
Caused by different structural modifications in the genome, fusion genes have gained attention because of their relationship with cancer.[63] The ability of RNA-seq to analyze a sample's whole transcriptome in an unbiased fashion makes it an attractive tool to find these kinds of common events in cancer.[64]
The idea follows from the process of aligning the short transcriptomic reads to a reference genome. Most of the short reads will fall within one complete exon, and a smaller but still large set would be expected to map to known exon-exon junctions. The remaining unmapped short reads would then be further analyzed to determine whether they match an exon-exon junction where the exons come from different genes. This would be evidence of a possible fusion event, however, because of the length of the reads, this could prove to be very noisy. An alternative approach is to use pair-end reads, when a potentially large number of paired reads would map each end to a different exon, giving better coverage of these events (see figure). Nonetheless, the end result consists of multiple and potentially novel combinations of genes providing an ideal starting point for further validation.
Application to genomic medicine
History
The past five years have seen a flourishing of NGS-based methods for genome analysis leading to the discovery of a number of new mutations and fusion transcripts in cancer. RNA-Seq data could help researchers interpreting the "personalized transcriptome" so that it will help understanding the transcriptomic changes happening therefore, ideally, identifying gene drivers for a disease. The feasibility of this approach is however dictated by the costs in terms of money and time.
A basic search on PubMed reveals that the term RNA Seq, queried as ""RNA Seq" OR "RNA-Seq" OR "RNA sequencing" OR "RNASeq"" in order to capture the most common ways of phrasing it, gives 5.425 hits demonstrating usage statistics of this technology. A few examples will be taken into consideration to explain that RNA-Seq applications to the clinic have the potentials to significantly affect patient's life and, on the other hand, requires a team of specialists (bioinformaticians, physicians/clinicians, basic researchers, technicians) to fully interpret the huge amount of data generated by this analysis.
As an example of clinical applications, researchers at the Mayo Clinic used an RNA-Seq approach to identify differentially expressed transcripts between oral cancer and normal tissue samples. They also accurately evaluated the allelic imbalance (AI), ratio of the transcripts produced by the single alleles, within a subgroup of genes involved in cell differentiation, adhesion, cell motility and muscle contraction[65] identifying a unique transcriptomic and genomic signature in oral cancer patients. Novel insight on skin cancer (melanoma) also come from RNA-Seq of melanoma patients. This approach led to the identification of eleven novel gene fusion transcripts originated from previously unknown chromosomal rearrangements. Twelve novel chimeric transcripts were also reported, including seven of those that confirmed previously identified data in multiple melanoma samples.[66] Furthermore, this approach is not limited to cancer patients. RNA-Seq has been used to study other important chronic diseases such as Alzheimer (AD) and diabetes. In the former case, Twine and colleagues compared the transcriptome of different lobes of deceased AD's patient's brain with the brain of healthy individuals identifying a lower number of splice variants in AD's patients and differential promoter usage of the APOE-001 and -002 isoforms in AD's brains.[67] In the latter case, different groups showed the unicity of the beta-cells transcriptome in diabetic patients in terms of transcripts accumulation and differential promoter usage[68] and long non coding RNAs (lncRNAs) signature.[69]
Compared with microarrays, NGS technology has identified novel and low frequency RNAs associated with disease processes. This advantage aids in the diagnosis and possible future treatments of diseases, including cancer. For example, NGS technology identified several previously undocumented differentially-expressed transcripts in rats treated with AFB1, a potent hepatocarcinogen. Nearly 50 new differentially-expressed transcriptions were identified between the controls and AFB1-treated rats. Additionally potential new exons were identified, including some that are responsive to AFB1. The next-generation sequencing pipeline identified more differential gene expressions compared with microarrays, particularly when DESeq software was utilized. Cufflinks identified two novel transcripts that were not previously annotated in the Ensembl database; these transcripts were confirmed using cloning PCR.[70] Numerous other studies have demonstrated NGS's ability to detect aberrant mRNA and small non-coding RNA expression in disease processes above that provided by microarrays. The lower cost and higher throughput offered by NGS confers another advantage to researchers.
The role of small non-coding RNAs in disease processes has also been explored in recent years. For example, Han et al. (2011) examined microRNA expression differences in bladder cancer patients in order to understand how changes and dysregulation in microRNA can influence mRNA expression and function. Several microRNAs were differentially expressed in the bladder cancer patients. Upregulation in the aberrant microRNAs was more common than downregulation in the cancer patients. One of the upregulated microRNAs, has-miR-96, has been associated with carcinogenesis, and several of the overexpressed microRNAs have also been observed in other cancers, including ovarian and cervical. Some of the downregulated microRNAs in cancer samples were hypothesized to have inhibitory roles.[71]
ENCODE and TCGA
A lot of emphasis has been given to RNA-Seq data after the Encyclopedia of DNA Elements (ENCODE) and The Cancer Genome Atlas (TCGA) projects have used this approach to characterize dozens of cell lines[72] and thousands of primary tumor samples,[73] respectively. ENCODE aimed to identify genome-wide regulatory regions in different cohort of cell lines and transcriptomic data are paramount in order to understand the downstream effect of those epigenetic and genetic regulatory layers. TCGA, instead, aimed to collect and analyze thousands of patient's samples from 30 different tumor types in order to understand the underlying mechanisms of malignant transformation and progression. In this context RNA-Seq data provide a unique snapshot of the transcriptomic status of the disease and look at an unbiased population of transcripts that allows the identification of novel transcripts, fusion transcripts and non-coding RNAs that could be undetected with different technologies.
See also
References
- 1 2 Ryan D. Morin; Matthew Bainbridge; Anthony Fejes; Martin Hirst; Martin Krzywinski; Trevor J. Pugh; Helen McDonald; Richard Varhol; Steven J.M. Jones & Marco A. Marra. (2008). "Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing". BioTechniques. 45 (1): 81–94. PMID 18611170. doi:10.2144/000112900.
- ↑ Chu Y, Corey DR (August 2012). "RNA sequencing: platform selection, experimental design, and data interpretation". Nucleic Acid Ther. 22 (4): 271–4. PMC 3426205
 . PMID 22830413. doi:10.1089/nat.2012.0367.
. PMID 22830413. doi:10.1089/nat.2012.0367. - ↑ Wang, Zhong; Gerstein, Mark; Snyder, Michael. "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews Genetics. 10 (1): 57–63. PMC 2949280 . PMID 19015660. doi:10.1038/nrg2484.
- ↑ Maher CA, Kumar-Sinha C, Cao X, et al. (March 2009). "Transcriptome sequencing to detect gene fusions in cancer". Nature. 458 (7234): 97–101. PMC 2725402 . PMID 19136943. doi:10.1038/nature07638.
- ↑ Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS (August 2012). "The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments". Nat Protoc. 7 (8): 1534–50. PMC 3535016 . PMID 22836135. doi:10.1038/nprot.2012.086.
- ↑ Kukurba, Kimberly R.; Montgomery, Stephen B. (2015-11-01). "RNA Sequencing and Analysis". Cold Spring Harbor Protocols. 2015 (11): 951–969. ISSN 1559-6095. PMC 4863231 . PMID 25870306. doi:10.1101/pdb.top084970.
- 1 2 3 Griffith, Malachi; Walker, Jason R.; Spies, Nicholas C.; Ainscough, Benjamin J.; Griffith, Obi L.; Ouellette, Francis (6 August 2015). "Informatics for RNA Sequencing: A Web Resource for Analysis on the Cloud". PLOS Computational Biology. 11 (8): e1004393. doi:10.1371/journal.pcbi.1004393.
- 1 2 Wang Z, Gerstein M, Snyder M (January 2009). "RNA-Seq: a revolutionary tool for transcriptomics". Nature Reviews Genetics. 10 (1): 57–63. PMC 2949280 . PMID 19015660. doi:10.1038/nrg2484.
- ↑ "RNA-seqlopedia". rnaseq.uoregon.edu. Retrieved 2017-02-08.
- 1 2 Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008). "Mapping and quantifying mammalian transcriptomes by RNA-seq". Nature Methods. 5 (7): 621–628. PMID 18516045. doi:10.1038/nmeth.1226.
- ↑ The Protocol Online websitehttp://www.protocol-online.org/prot/Molecular_Biology/RNA/RNA_Extraction/mRNA_Isolation/index.html provides a list of several protocols relating to mRNA isolation
- ↑ Chen, Emily A; Souaiaia, Tade; Herstein, Jennifer S; Evgrafov, Oleg V; Spitsyna, Valeria N; Rebolini, Danea F; Knowles, James A (2014). "Effect of RNA integrity on uniquely mapped reads in RNA-Seq". BMC Research Notes. 7 (1): 753. doi:10.1186/1756-0500-7-753.
- ↑ Liu D, Graber JH (2006). "Quantitative comparison of EST libraries requires compensation for systematic biases in cDNA generation". BMC Bioinformatics. 7: 77. PMC 1431573 . PMID 16503995. doi:10.1186/1471-2105-7-77.
- 1 2 Li H, Lovci MT, Kwon YS, Rosenfeld MG, Fu XD, Yeo GW (2008). "Determination of tag density required for digital transcriptome analysis: Application to an androgen-sensitive prostate cancer model". Proc Natl Acad Sci USA. 105 (51): 20179–84. PMC 2603435 . PMID 19088194. doi:10.1073/pnas.0807121105.
- ↑ Stegle, Oliver; Parts, Leopold; Piipari, Matias; Winn, John; Durbin, Richard (16 February 2012). "Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses". Nature Protocols. 7 (3): 500–507. PMC 3398141 . PMID 22343431. doi:10.1038/nprot.2011.457.
- ↑ Kingsford, Carl; Patro, Rob (15 June 2015). "Reference-based compression of short-read sequences using path encoding". Bioinformatics. 31 (12): 1920–1928. PMC 4481695 . PMID 25649622. doi:10.1093/bioinformatics/btv071.
- 1 2 Grabherr, Manfred G; Haas, Brian J; Yassour, Moran; Levin, Joshua Z; Thompson, Dawn A; Amit, Ido; Adiconis, Xian; Fan, Lin; Raychowdhury, Raktima; Zeng, Qiandong; Chen, Zehua; Mauceli, Evan; Hacohen, Nir; Gnirke, Andreas; Rhind, Nicholas; di Palma, Federica; Birren, Bruce W; Nusbaum, Chad; Lindblad-Toh, Kerstin; Friedman, Nir; Regev, Aviv (15 May 2011). "Full-length transcriptome assembly from RNA-Seq data without a reference genome". Nature Biotechnology. 29 (7): 644–652. PMC 3571712 . PMID 21572440. doi:10.1038/nbt.1883.
- ↑ "De Novo Assembly Using Illumina Reads" (PDF). Retrieved 22 October 2016.
- ↑ Zerbino, D. R.; Birney, E. (21 February 2008). "Velvet: Algorithms for de novo short read assembly using de Bruijn graphs". Genome Research. 18 (5): 821–829. PMC 2336801 . PMID 18349386. doi:10.1101/gr.074492.107.
- ↑ Oases: a transcriptome assembler for very short reads
- ↑ Chang, Zheng; Li, Guojun; Liu, Juntao; Zhang, Yu; Ashby, Cody; Liu, Deli; Cramer, Carole L; Huang, Xiuzhen (2015). "Bridger: a new framework for de novo transcriptome assembly using RNA-seq data". Genome Biology. 16 (1): 30. PMC 4342890 . PMID 25723335. doi:10.1186/s13059-015-0596-2.
- 1 2 Li, Bo; Fillmore, Nathanael; Bai, Yongsheng; Collins, Mike; Thomson, James A; Stewart, Ron; Dewey, Colin N (21 December 2014). "Evaluation of de novo transcriptome assemblies from RNA-Seq data". Genome Biology. 15 (12). doi:10.1186/s13059-014-0553-5.
- 1 2 Dobin, A.; Davis, C. A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T. R. (25 October 2012). "STAR: ultrafast universal RNA-seq aligner". Bioinformatics. 29 (1): 15–21. PMC 3530905 . PMID 23104886. doi:10.1093/bioinformatics/bts635.
- ↑ Langmead, Ben; Trapnell, Cole; Pop, Mihai; Salzberg, Steven L (2009). "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome". Genome Biology. 10 (3): R25. PMC 2690996 . PMID 19261174. doi:10.1186/gb-2009-10-3-r25.
- ↑ Trapnell, C.; Pachter, L.; Salzberg, S. L. (16 March 2009). "TopHat: discovering splice junctions with RNA-Seq". Bioinformatics. 25 (9): 1105–1111. PMC 2672628 . PMID 19289445. doi:10.1093/bioinformatics/btp120.
- ↑ Trapnell, Cole; Roberts, Adam; Goff, Loyal; Pertea, Geo; Kim, Daehwan; Kelley, David R; Pimentel, Harold; Salzberg, Steven L; Rinn, John L; Pachter, Lior (1 March 2012). "Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks". Nature Protocols. 7 (3): 562–578. PMC 3334321 . PMID 22383036. doi:10.1038/nprot.2012.016.
- ↑ Liao, Y.; Smyth, G. K.; Shi, W. (4 April 2013). "The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote". Nucleic Acids Research. 41 (10): e108–e108. PMC 3664803 . PMID 23558742. doi:10.1093/nar/gkt214.
- ↑ Patro, Rob; Mount, Stephen M; Kingsford, Carl (20 April 2014). "Sailfish enables alignment-free isoform quantification from RNA-seq reads using lightweight algorithms". Nature Biotechnology. 32 (5): 462–464. PMC 4077321 . PMID 24752080. doi:10.1038/nbt.2862.
- ↑ Bray, Nicolas L; Pimentel, Harold; Melsted, Páll; Pachter, Lior (4 April 2016). "Near-optimal probabilistic RNA-seq quantification". Nature Biotechnology. 34 (5): 525–527. doi:10.1038/nbt.3519.
- ↑ Wu, TD; Watanabe, CK (1 May 2005). "GMAP: a genomic mapping and alignment program for mRNA and EST sequences.". Bioinformatics (Oxford, England). 21 (9): 1859–75. PMID 15728110. doi:10.1093/bioinformatics/bti310.
- ↑ Lu B, Zeng Z, Shi T (February 2013). "Comparative study of de novo assembly and genome-guided assembly strategies for transcriptome reconstruction based on RNA-Seq". Science China Life Sciences. 56 (2): 143–55. PMID 23393030. doi:10.1007/s11427-013-4442-z.
- ↑ Bradnam KR, Fass JN, Alexandrov A, et al. (July 2013). "Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species". Gigascience. 2 (1): 10. PMC 3844414 . PMID 23870653. doi:10.1186/2047-217X-2-10.
- ↑ Greenbaum D, Colangelo C, Williams K, Gerstein M (2003). "Comparing protein abundance and mRNA expression levels on a genomic scale". Genome Biology. 4 (9): 117. PMC 193646 . PMID 12952525. doi:10.1186/gb-2003-4-9-117.
- ↑ Zhang, Zong Hong; Jhaveri, Dhanisha J.; Marshall, Vikki M.; Bauer, Denis C.; Edson, Janette; Narayanan, Ramesh K.; Robinson, Gregory J.; Lundberg, Andreas E.; Bartlett, Perry F.; Wray, Naomi R.; Zhao, Qiong-Yi; Provero, Paolo (13 August 2014). "A Comparative Study of Techniques for Differential Expression Analysis on RNA-Seq Data". PLoS ONE. 9 (8): e103207. doi:10.1371/journal.pone.0103207.
- ↑ Anders, S; Pyl, PT; Huber, W (15 January 2015). "HTSeq--a Python framework to work with high-throughput sequencing data.". Bioinformatics (Oxford, England). 31 (2): 166–9. PMC 4287950 . PMID 25260700. doi:10.1093/bioinformatics/btu638.
- ↑ Liao, Y; Smyth, GK; Shi, W (1 April 2014). "featureCounts: an efficient general purpose program for assigning sequence reads to genomic features.". Bioinformatics (Oxford, England). 30 (7): 923–30. PMID 24227677. doi:10.1093/bioinformatics/btt656.
- ↑ Schmid, MW; Grossniklaus, U (1 February 2015). "Rcount: simple and flexible RNA-Seq read counting.". Bioinformatics (Oxford, England). 31 (3): 436–7. PMID 25322836. doi:10.1093/bioinformatics/btu680.
- ↑ Finotello, F; Lavezzo, E; Bianco, L; Barzon, L; Mazzon, P; Fontana, P; Toppo, S; Di Camillo, B (2014). "Reducing bias in RNA sequencing data: a novel approach to compute counts.". BMC Bioinformatics. 15 Suppl 1: S7. PMC 4016203 . PMID 24564404. doi:10.1186/1471-2105-15-s1-s7.
- ↑ Hashimoto, TB; Edwards, MD; Gifford, DK (6 March 2014). "Universal count correction for high-throughput sequencing.". PLOS Computational Biology. 10 (3): e1003494. PMC 3945112 . PMID 24603409. doi:10.1371/journal.pcbi.1003494.
- ↑ Robinson, Mark D; Oshlack, Alicia (2010). "A scaling normalization method for differential expression analysis of RNA-seq data". Genome Biology. 11 (3): R25. PMC 2864565 . PMID 20196867. doi:10.1186/gb-2010-11-3-r25.
- ↑ Trapnell, Cole; Williams, Brian A; Pertea, Geo; Mortazavi, Ali; Kwan, Gordon; van Baren, Marijke J; Salzberg, Steven L; Wold, Barbara J; Pachter, Lior (May 2010). "Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation". Nat Biotechnol. 28 (5): 511–515. PMC 3146043 . PMID 20436464. doi:10.1038/nbt.1621.
- ↑ Robinson, Mark D; Oshlack, Alicia (2010). "A scaling normalization method for differential expression analysis of RNA-seq data". Genome Biology. 11 (3): R25. PMC 2864565 . PMID 20196867. doi:10.1186/gb-2010-11-3-r25.
- ↑ Law, Charity W; Chen, Yunshun; Shi, Wei; Smyth, Gordon K (2014). "voom: precision weights unlock linear model analysis tools for RNA-seq read counts". Genome Biology. 15 (2): R29. PMC 4053721 . PMID 24485249. doi:10.1186/gb-2014-15-2-r29.
- ↑ Anders, Simon; Huber, Wolfgang (2010). "Differential expression analysis for sequence count data". Genome Biology. 11 (10): R106. PMC 3218662 . PMID 20979621. doi:10.1186/gb-2010-11-10-r106.
- ↑ Robinson, MD; McCarthy, DJ; Smyth, GK (1 January 2010). "edgeR: a Bioconductor package for differential expression analysis of digital gene expression data.". Bioinformatics (Oxford, England). 26 (1): 139–40. PMC 2796818 . PMID 19910308. doi:10.1093/bioinformatics/btp616.
- ↑ Soneson, Charlotte; Delorenzi, Mauro (2013-01-01). "A comparison of methods for differential expression analysis of RNA-seq data". BMC Bioinformatics. 14: 91. ISSN 1471-2105. PMC 3608160 . PMID 23497356. doi:10.1186/1471-2105-14-91.
- 1 2 Anders, Simon; Huber, Wolfgang (2010-01-01). "Differential expression analysis for sequence count data". Genome Biology. 11 (10): R106. ISSN 1474-760X. PMC 3218662 . PMID 20979621. doi:10.1186/gb-2010-11-10-r106.
- 1 2 Robinson, Mark D.; McCarthy, Davis J.; Smyth, Gordon K. (2010-01-01). "edgeR: a Bioconductor package for differential expression analysis of digital gene expression data". Bioinformatics (Oxford, England). 26 (1): 139–140. ISSN 1367-4811. PMC 2796818 . PMID 19910308. doi:10.1093/bioinformatics/btp616.
- ↑ "Bioconductor - Open source software for bioinformatics".
- ↑ Huber, Wolfgang; Carey, Vincent J; Gentleman, Robert; Anders, Simon; Carlson, Marc; Carvalho, Benilton S; Bravo, Hector Corrada; Davis, Sean; Gatto, Laurent. "Orchestrating high-throughput genomic analysis with Bioconductor". Nature Methods. 12 (2): 115–121. PMC 4509590 . PMID 25633503. doi:10.1038/nmeth.3252.
- ↑ Mortazavi, Ali; Williams, Brian A.; McCue, Kenneth; Schaeffer, Lorian; Wold, Barbara (2008-07-01). "Mapping and quantifying mammalian transcriptomes by RNA-Seq". Nature Methods. 5 (7): 621–628. ISSN 1548-7105. PMID 18516045. doi:10.1038/nmeth.1226.
- ↑ Marguerat, Samuel; Schmidt, Alexander; Codlin, Sandra; Chen, Wei; Aebersold, Ruedi; Bähler, Jürg (2012-10-26). "Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells". Cell. 151 (3): 671–683. ISSN 1097-4172. PMC 3482660 . PMID 23101633. doi:10.1016/j.cell.2012.09.019.
- ↑ Owens, Nick D. L.; Blitz, Ira L.; Lane, Maura A.; Patrushev, Ilya; Overton, John D.; Gilchrist, Michael J.; Cho, Ken W. Y.; Khokha, Mustafa K. (2016-01-26). "Measuring Absolute RNA Copy Numbers at High Temporal Resolution Reveals Transcriptome Kinetics in Development". Cell Reports. 14 (3): 632–647. ISSN 2211-1247. PMC 4731879 . PMID 26774488. doi:10.1016/j.celrep.2015.12.050.
- 1 2 Marcotte, EM.; Pellegrini, M.; Thompson, MJ.; Yeates, TO.; Eisenberg, D. (Nov 1999). "A combined algorithm for genome-wide prediction of protein function.". Nature. 402 (6757): 83–6. PMID 10573421. doi:10.1038/47048.
- 1 2 Giorgi Federico Manuel (2013). "Comparative study of RNA-seq- and Microarray-derived coexpression networks in Arabidopsis thaliana". Bioinformatics. 29 (6): 717–724. PMID 23376351. doi:10.1093/bioinformatics/btt053.
- ↑ Iancu Ovidiu D (2012). "Utilizing RNA-Seq data for de novo coexpression network inference". Bioinformatics. 28 (12): 1592–1597. PMC 3493127 . PMID 22556371. doi:10.1093/bioinformatics/bts245.
- ↑ Eksi, R; Li, HD; Menon, R; Wen, Y; Omenn, GS; Kretzler, M; Guan, Y (Nov 2013). "Systematically differentiating functions for alternatively spliced isoforms through integrating RNA-seq data.". PLOS Computational Biology. 9 (11): e1003314. PMC 3820534 . PMID 24244129. doi:10.1371/journal.pcbi.1003314.
- ↑ Li, HD; Menon, R; Omenn, GS; Guan, Y (Jun 17, 2014). "The emerging era of genomic data integration for analyzing splice isoform function.". Trends in genetics : TIG. 30 (8): 340–347. PMID 24951248. doi:10.1016/j.tig.2014.05.005.
- ↑ Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS (2007). "SNP discovery via 454 transcriptome sequencing". The Plant Journal. 51 (5): 910–918. PMC 2169515 . PMID 17662031. doi:10.1111/j.1365-313X.2007.03193.x.
- ↑ Lalonde E, Ha KC, Wang Z, et al. (April 2011). "RNA sequencing reveals the role of splicing polymorphisms in regulating human gene expression". Genome Res. 21 (4): 545–54. PMC 3065702 . PMID 21173033. doi:10.1101/gr.111211.110.
- ↑ "CummeRbund - An R package for persistent storage, analysis, and visualization of RNA-Seq from cufflinks output". Retrieved 2013-07-28.
- ↑ Garcion E, Wallace B, Pelletier L, Wion D (2004). "RNA mutagenesis and sporadic prion diseases". Journal of Theoretical Biology. 230 (2): 271–274. PMID 15302558. doi:10.1016/j.jtbi.2004.05.014.
- ↑ Teixeira MR (2006). "Recurrent fusion oncogenes in carcinomas". Ciritical Reviews in Oncogenesis. 12 (3–4): 257–271. PMID 17425505. doi:10.1615/critrevoncog.v12.i3-4.40.
- ↑ Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, Sam L, Barrette T, Palanisamy N, Chinnaiyan AM (January 2009). "Transcriptome Sequencing to Detect Gene Fusions in Cancer". Nature. 458 (7234): 97–101. PMC 2725402 . PMID 19136943. doi:10.1038/nature07638.
- ↑ Tuch BB, Laborde RR, Xu X, et al. (2010). "Tumor transcriptome sequencing reveals allelic expression imbalances associated with copy number alterations". PLoS ONE. 5 (2): e9317. PMC 2824832 . PMID 20174472. doi:10.1371/journal.pone.0009317.
- ↑ Berger MF, Levin JZ, Vijayendran K, et al. (April 2010). "Integrative analysis of the melanoma transcriptome". Genome Res. 20 (4): 413–27. PMC 2847744 . PMID 20179022. doi:10.1101/gr.103697.109.
- ↑ Twine NA, Janitz K, Wilkins MR, Janitz M (2011). "Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer's disease". PLoS ONE. 6 (1): e16266. PMC 3025006 . PMID 21283692. doi:10.1371/journal.pone.0016266.
- ↑ Ku GM, Kim H, Vaughn IW, et al. (October 2012). "Research resource: RNA-Seq reveals unique features of the pancreatic β-cell transcriptome". Mol. Endocrinol. 26 (10): 1783–92. PMC 3458219 . PMID 22915829. doi:10.1210/me.2012-1176.
- ↑ Morán I, Akerman I, van de Bunt M, et al. (October 2012). "Human β cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes". Cell Metab. 16 (4): 435–48. PMC 3475176 . PMID 23040067. doi:10.1016/j.cmet.2012.08.010.
- ↑ Merrick B. A.; Phadke D. P.; Auerbach S. S.; Mav D.; Stiegelmeyer S. M.; Shah R. R.; Tice R. R. (2013). "RNA-seq reveals novel hepatic gene expression pattern in Aflatoxin B1 treated rats". PLoS ONE. 8: e61768. PMC 3632591 . PMID 23630614. doi:10.1371/journal.pone.0061768.
- ↑ Han Y.; Chen J.; Zhao X.; Liang C.; Wang Y.; Sun L.; Jiang Z.; Zhang Z.; Yang R.; Chen J.; Li Z.; Tang A.; Li X.; Ye J.; Guan Z.; Gui Y.; Cai Z. (2011). "MicroRNA expression signatures of bladder cancer revealed by deep sequencing". PLOS ONE. 6: e18286. PMC 3065473 . PMID 21464941. doi:10.1371/journal.pone.0018286.
- ↑ "ENCODE Data Matrix". Retrieved 2013-07-28.
- ↑ "The Cancer Genome Atlas - Data Portal". Retrieved 2013-07-28.
External links
- RNA-Seq for Everyone: a high-level guide to designing and implementing an RNA-Seq experiment.