Nutriepigenomics
Nutriepigenomics is the study of food nutrients and their effects on human health through epigenetic modifications. There is now considerable evidence that nutritional imbalances during gestation and lactation are linked to non-communicable diseases, such as obesity, cardiovascular disease, diabetes, hypertension, and cancer. If metabolic disturbances occur during critical time windows of development, the resulting epigenetic alterations can lead to permanent changes in tissue and organ structure or function and predispose individuals to disease.[1]
Overview
Epigenetics relates to heritable changes in gene function that occur independently of alterations in primary DNA sequence. Two major epigenetic mechanisms implicated in nutriepigenomics are DNA methylation and histone modification. DNA methylation in gene promoter regions usually results in gene silencing and influences gene expression. While this form of gene silencing is extremely important in development and cellular differentiation, aberrant DNA methylation can be detrimental and has been linked to various disease processes, such as cancer.[2] The methyl groups used in DNA methylation are often derived from dietary sources, such as folate and choline, and explains why diet can have a significant impact on methylation patterns and gene expression.[3] Gene silencing can also be reinforced through the recruitment of histone deacetylases to decrease transcriptional activation. Conversely, histone acetylation induces transcriptional activation to increase gene expression. Dietary components can influence these epigenetic events, thereby altering gene expression and disturbing functions such as appetite control, metabolic balance and fuel utilization.[1]
Various genetic sequences can be targeted for epigenetic modification. A transcriptome-wide analysis in mice found that a protein-restricted (PR) diet during gestation resulted in differential gene expression in approximately 1% of the fetal genes analyzed (235/22,690). Specifically, increased expression was seen in genes involved in the p53 pathway, apoptosis, negative regulators of cell metabolism, and genes related to epigenetic control.[4] Additional studies have investigated the effect of a PR-diet in rats and found changes in promoter methylation of both the glucocorticoid receptor and peroxisome proliferator-activated receptor (PPAR).[5][6] Altered expression of these receptors can result in elevated blood glucose levels and affect lipid and carbohydrate metabolism.[3] Feeding a PR-diet to pregnant and/or lactating mice also increased expression of glucokinase, acetyl-CoA carboxylase, PPARα, and acyl-CoA oxidase.[7] Changes in expression were reportedly due to epigenetic regulation of either the gene promoter itself, or promoters of transcription factors that regulate gene expression. Additional genes that have been shown, either by in vitro or in vivo studies, to be regulated by epigenetic mechanisms include leptin, SOCS3, glucose transporter (GLUT)-4, POMC, 11-β-hydroxysteroid dehydrogenase type 2 and corticotrophin releasing hormone. Epigenetic modification of these genes may lead to “metabolic programming” of the fetus and result in long-term changes in metabolism and energy homeostasis.[8]
Nutriepigenomics and development
The period of development in which the nutritional imbalance occurs is very important in determining which disease-related genes will be affected. Different organs have critical developmental stages, and the time point at which they are compromised will predispose individuals to specific diseases.[9] Epigenetic modifications that occur during development may not be expressed until later in life depending on the function of the gene.[3] While the majority of studies implicate prenatal and perinatal periods as critical time windows, some research has shown that nutritional intake during adulthood can also affect the epigenome.
Prenatal
Developmental plasticity is the process in which fetuses adapt to their environment. Environmental cues, including dietary components, present in the in utero environment can induce significant changes in the expression of the genome through epigenetic modifications.[7] Fetal developmental plastic responses can cause changes in lean body mass, endocrinology, blood flow and vascular loading, and lead to increased risk of various diseases in adulthood.
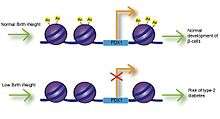
Low Birth Weight
Fetal exposure to calcium, folate, magnesium, high or low protein, and zinc have all been associated with birth weight.[9] Numerous studies have investigated the link between birth weight and risk of disease and have found that low birth weight is significantly associated with coronary heart disease, stroke and type-2 diabetes. Most importantly, these associations occurred after adjusting for lifestyle factors, implying a genetic basis for onset of disease.[10] Impaired insulin secretion is associated with low birth weight and can lead to insulin resistance as babies accumulate body fat.[11] Studies using intrauterine growth retarded (IUGR) rats have found that growth inhibition can lead to decreased expression of Pdx1 transcription factor, which is essential for differentiation and function of pancreatic beta cells.[12] Decreased histone acetylation at the proximal promoter of Pdx1 is responsible for reduced Pdx1 expression and subsequently results in a cascade of histone deacetylation and methylation events that can result in type-2 diabetes.
Obesity
Obesity during pregnancy and high-fat maternal diets both show strong associations with obesity in offspring. As the number of overweight reproductive-age women increases, the number of overweight children and infants also increases.[10] It has been postulated that maternal obesity causes an accumulation of fat in fetal adipose tissue (adiposity) and predisposes babies for obesity in childhood and adulthood.[10] Animal studies have shown that maternal overnutrition may impact brain development and cause disruptions to programming of the hypothalamus. Offspring that were exposed to a high-fat or high-caloric maternal diet had increased levels of insulin, glucose and leptin. It is hypothesized that these elevations are due to disturbances in the complex neuronal network that includes the neuropeptide Y (NPY) and proopiomelanocortin (POMC) pathways.[8] This altered neuronal signaling can consequently impact food-intake behavior and lead to diet-induced obesity in adulthood. While epigenetic modifications are most likely involved in the development of obesity, the specific target genes have yet to be identified. Genes involved in adipogenesis, such as fibroblast growth-factor-2, phosphatase and tensin homologue, cyclin-dependent kinase inhibitor 1A and oestrogen receptor-alpha, possess multiple CpG islands in their promoter sites and may act as epigenetic targets.[13] Furthermore, it has been shown that prenatal exposure to a hypomethylating agent, such as bisphenol A (BPA), is associated with increased body weight and suggests modified DNA methylation as a mechanism for increasing susceptibility to obesity.[13]
Folate
It has long been realized that maternal folate intake during pregnancy is linked to fetal development and growth, and can reduce the risk of serious birth defects. Folate is a source of S-adenosyl methionine (SAM), which is used to supply DNA methyltransferases with methyl groups. Therefore, changes in folate supply have a substantial effect on DNA methylation patterns. Low levels of folate are associated with an increased risk of preterm delivery, poor growth of the placenta and uterus, and intrauterine growth retardation.[3] Several complex diseases, including cancer, cardiovascular diseases and autism have also been linked to maternal folate status. Based on animal studies it has been hypothesized that reduced folate intake could increase the risk of neural tube defects by reducing the amount of methylayed DNA during cranial neural tube closure.[14] Recently it was discovered that folate protection from congenital heart defects is linked to epigenetics and Wnt signaling. Multiple environmental factors target the Wnt signaling pathway during embryogenesis and can cause misregulation of the pathway. Folic acid metabolism generates SAM, thereby altering the methylation states of histones H3K9, H3K4, and H3K27 and genetically altering Wnt signaling.[15]
Recently a double-blind placebo controlled trial of high dose Folinic Acid (Leucovorin Calcium) demonstrated efficacy at improving verbal communication in children with autism.[16]
Perinatal
Another critical developmental time window is the perinatal period, the time period immediately before and after birth. It has been shown that maternal diet in late pregnancy and an infant’s diet in the beginning weeks can all have significant impacts on gene expression. Therefore, perinatal nutrition is both late-stage in utero nutrition and lactation.
Bone health
Bone mass and the development of osteoporosis have been studied in relation to perinatal nutrition. An important factor to consider when investigating perinatal nutrition is whether the baby was breast-fed or formula-fed. Studies have shown that breast-fed babies have increased bone mass compared to those were not breast-fed, and that this small increase in bone mass during a period of critical development could potentially program the skeleton to continue along a “healthy” growth trajectory.[17] It has also been shown that maternal vitamin D insufficiency during late pregnancy is associated with reduced bone size and mineral mass in late childhood.[18] Peak bone mass has shown to be a good predictor of risk of fracture and osteoporosis, with even a small increase in peak bone mass resulting in a much lower risk of bone fracture.[9] Research shows that genetic markers explain only a small proportion of variation in bone mass and risk of fracture. Therefore, healthy bone programming is most likely influenced by various epigenetic mechanisms, such as imprinting of the growth promoting genes IGF-2, or changes to the hypothalamic-pituitary-adrenal axis (HPA).[19]
Neurodevelopment
Imbalances in maternal nutrition can also have a significant effect on fetal neurodevelopment. Brain development occurs most rapidly during fetal development and infancy, and research has shown that exposure to certain environmental conditions can have long-lasting effects on cognition. Specifically, n-3 fatty acids, iodine, iron and choline have been shown to influence brain development and impact cognitive ability and behavior. The greatest evidence for a link between nutrition and neurodevelopment comes from studies that show low birth weight associated with low IQ and increased risk of schizophrenia.[20][21] Several studies suggest that breast-feeding promotes long-term neurodevelopment by providing the nutrients necessary for proper brain development.[22] A study in mice showed that choline-deficient diets during the late gestation period impaired fetal brain development, including decreased cell proliferation and reduced visual-spatial and auditory memory.[23] These cognitive changes appeared to be due to altered histone and DNA methylation patterns in the fetal hippocampus, thus providing a link between maternal nutrition, epigenetics, and early brain development.
Type-1 diabetes
It has been postulated that breast-feeding may also protect against type-1 diabetes, with research showing that formula-fed infants are at an increased risk of developing islet autoantibodies. Individuals with type-1 diabetes experience a pre-clinical diabetes phase characterized by autoimmunity against pancreatic islets.[24] The introduction of certain foods in the first few months of life, such as berries and cereal, is significantly associated with increased risk of islet autoantibody development compared to babies who are exposed to solid foods later in life.[25] While the pathogenesis behind development of autoantibodies remains largely unknown, it is very probable that an epigenetic link exists between perinatal diet and risk of type-1 diabetes.[9]
Adulthood
The majority of research in nutriepigenomics has focused on nutritional imbalances during gestation and lactation periods. However, foods that are consumed during adulthood can also impact gene expression and disease pathogenesis. Cancer is the disease most commonly associated with adult nutrition and epigenetic modifications. DNA hypomethelation promotes cancer progression by allowing increased gene transcription, while hypermethylation can silence tumor suppressor genes and further promote uncontrolled cell division and tumor formation. Compounds found in foods, such as genistein and tea polyphenols, are able to regulate DNA methyltransferases and histone acetylation in cultured cancer cells and may provide protection against certain types of cancer.[13] Other dietary compounds, such as diallyl disulfide present in garlic and sulforaphane present in cruciferous vegetables, have been associated with cancer prevention in clinical trials.[26] This may be due to their ability to inhibit histone deacetylase (HDAC) enzymes and prevent silencing of important regulatory genes.
Transgenerational effects
Many believe epigenetic regulation is cleared during the fertilization process, yet more evidence for transgenerational effects (TGEs) are being revealed.[1] These TGEs take place when the epigenetic regulatory patterns are not sufficiently erased during fertilization, possibly due to nutrition levels in previous generations. Later generations may be affected from caloric and protein restriction, high-fat interventions and endocrine disruption in earlier generations.[1] Differences within the nutritional behavior of the maternal rat are believed to cause malprogramming in the F1 generation and may then be passed to subsequent generations.[1] Maternal rats fed a PR-diet during the entire length of pregnancy led to metabolic-related problems in the F1 and F2 generations, even with normal nutrition during the F1 pregnancy.[27][28] These effects have also been seen in the F3 generation depending on the length of protein restriction.[29][30] If protein restriction occurred solely during pregnancy, the F1 and F2 offspring had higher systolic blood pressure and lower nephron numbers, possibly predisposing them to hypertension.[30] Altered glucose utilization was detected in the grand-offspring of maternal rats fed a PR-diet during pregnancy and lactation, potentially resulting in diabetes later on in life [29]
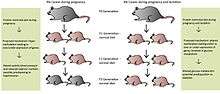
Protein-restriction in the F0 generation led to hypomethylation of promoters involved in metabolism in the F1 and F2 generations, even though the F1 pregnant rat was given a normal diet.[31] The exact mechanism of this situation has yet to be elucidated; however, direct transmission is a distinct possibility, meaning the epigenetic marks were preserved during spermatogenesis and oogenesis, when they are normally erased.
Models used in nutriepigenomic studies
Most research to date use common rodent models to investigate the role of nutrition on phenotype.[10] Popular areas to investigate include IUGR studies, whereby rodents, and sometimes sheep, are subjected to a variety of nutritional conditions. A model for studying IUGR in rodent was developed by Simmons et al. (2010) and is used to investigate type II diabetes.[32] The maternal rats have their uterine arteries ligated, causing altered use of glucose and insulin in the fetus and can therefore serve as a model for diabetes. These growth-retarded rats were found to be highly similar to human fetuses, as they both display symptoms such as lowered glucose and insulin levels. Gestational diabetes may also be studied through chemical induction using streptozotocin treatment of the pregnant rats.[33] Streptozotocin can cause destruction of the beta cells within the pancreas depending on the concentration given.
The predominant means of investigating nutriepigenetics involves varying the nutritional conditions to which a subject is exposed to and monitoring the effects thereafter. Restricting caloric and protein intake are the two most common methods.[33] A pregnant rodent may have their caloric intake reduced up to 30-50% of normal intake. Protein restricted rodents are given 8-9% casein, as opposed to control rats that are fed 20% casein. Micronutrients, such as zinc and iron, may also be restricted to investigate the effects on offspring. Additionally, rats fed diets lacking or including methyl donors are often used to study the effects of diet on epigenomics, as variations within the methylation of DNA are common means of silencing or expressing genes.[34] Supplementing maternal mice with folic acid, vitamin B12, choline and betaine leads to increased levels of DNA methylation at CpG sites and causes a coat color change.[35] This is an example of epigenetically modifiable loci called a “metastable epiallele”, of which only a few have been identified. The above is an example of the “agouti” gene locus, whereby the insertion of a transposable element upstream to the Agouti gene is hypermethylated from the supplementation and causes a change in the mice's coat color. Diets containing higher carbohydrate and fat content attempt to mimic typical Western-style diets may also be used in nutriepigenetic studies.[10][33] Another method used is “catch-up”, where offspring of rats born to mothers subjected to various diets are subsequently cross-fostered to mothers fed normal diets.[33]
Future directions
The possibilities of utilizing nutriepigenomics for intervention are quite expansive. This can include preventative therapies, such as providing an optimal regime for nutrition during pregnancy and lactation.[33] It is already common place for pregnant mothers to supplement their diets with choline and folate to prevent the development of neurological disabilities in the fetus.
A highly specific diet, termed an "EpiG diet," may be employed for an individual believed to be at higher risk of developing a metabolic disorder.[1] These diets may include supplementation with methyl donors, such as folate. There are also many other natural compounds, such as resveratrol, curcumin and green tea that have been termed “epigenetic modifiers”, as they have anti-cancer capabilities in addition to being used as treatments for metabolic diseases.[36] However, the functions of these compounds still require long-term studies to evaluate their effect over time.
There also exists potential for therapeutic treatments that may correct metabolic disorders, such as type II diabetes.[33] Components of garlic and cruciferous vegetables are known to possess HDAC inhibitors that modify the acetylation of histone proteins and may contain a protection against cancer.[26] These same compounds have also been implicated in irritable bowel syndrome (IBS) and colon cancer, as they may modify the histones normally implicated in these diseases.[37]
Elucidation of disease pathways is another future direction for nutriepigenomic studies. For example, choline-deficient diets and alcohol metabolism during pregnancy may have very similar metabolic pathways.[38] Therefore, animal studies using choline-restricted diets may assist in investigations of fetal alcohol spectrum disorders.
When compared to studies of maternal transmission, investigations into the role of paternal diets are lacking. A review demonstrated the nutrition of both parents do in fact play a role in determining the health of their offspring.[39] A germ-line study reported paternal rats fed a high-fat diet led to insulin dysfunction in the F1 offspring.[40] While this likely occurs via epigenetic modifications similar to those postulated in the maternal diets, the exact mechanism remains to be defined. Assessing the role of epigenetic mechanisms may be easier using paternal inheritance, as sperm transmits epigenetic and genetic information, whereas the female cells also transmit mitochondrial DNA.[39]
See also
- Epigenetics
- Epigenome
- Epigenomics
- Genome
- Molecular epidemiology
- Molecular pathological epidemiology
- Nutritional epidemiology
Notes
- 1 2 3 4 5 6 Gallou-Kabani C, Vige A, Gross MS, Junien C. Nutri-epigenomics: Lifelong remodelling of our epigenomes by nutritional and metabolic factors and beyond. Clin Chem Lab Med. 2007;45(3):321-7 doi:10.1515/CCLM.2007.081 PMID 17378726
- ↑ Berdasco M, Esteller M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev Cell. 2010 Nov 16;19(5):698-711 doi:10.1016/j.devcel.2010.10.005 PMID 21074720.
- 1 2 3 4 Pozharny Y, Lambertini L, Clunie G, Ferrara L, Lee MJ. Epigenetics in women's health care. Mt Sinai J Med. 2010 Mar;77(2):225-35 doi:10.1002/msj.20176 PMID 20309920.
- ↑ Gheorghe CP, Goyal R, Holweger JD, Longo LD. Placental gene expression responses to maternal protein restriction in the mouse. Placenta. 2009 May;30(5):411-7 doi:10.1016/j.placenta.2009.03.002 PMID 19362366
- ↑ Lillycrop KA, Phillips ES, Torrens C, Hanson MA, Jackson AA, Burdge GC. Feeding pregnant rats a protein-restricted diet persistently alters the methylation of specific cytosines in the hepatic PPAR alpha promoter of the offspring. Br J Nutr. 2008 Aug;100(2):278-82 doi:10.1017/S0007114507894438 PMID 18186951
- ↑ Lillycrop KA, Slater-Jefferies JL, Hanson MA, Godfrey KM, Jackson AA, Burdge GC. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007 Jun;97(6):1064-73 doi:10.1017/S000711450769196X PMID 17433129
- 1 2 Burdge GC, Lillycrop KA. Nutrition, epigenetics, and developmental plasticity: Implications for understanding human disease. Annu Rev Nutr. 2010 Aug 21;30:315-39 doi:10.1146/annurev.nutr.012809.104751 PMID 20415585
- 1 2 Tamashiro KL, Moran TH. Perinatal environment and its influences on metabolic programming of offspring. Physiol Behav. 2010 Jul 14;100(5):560-6 doi:10.1016/j.physbeh.2010.04.008 PMID 20394764
- 1 2 3 4 Hanley B, Dijane J, Fewtrell M, Grynberg A, Hummel S, Junien C, Koletzko B, Lewis S, Renz H, Symonds M, Gros M, Harthoorn L, Mace K, Samuels F, van Der Beek EM. Metabolic imprinting, programming and epigenetics - a review of present priorities and future opportunities. Br J Nutr. 2010 Jul;104 Suppl 1:S1-25 doi:10.1017/S0007114510003338 PMID 20929595
- 1 2 3 4 5 Simmons R. Epigenetics and maternal nutrition: Nature v. nurture. Proc Nutr Soc. 2010 Nov 29:1-9 PMID 21110912
- ↑ Jensen CB, Storgaard H, Dela F, Holst JJ, Madsbad S, Vaag AA. Early differential defects of insulin secretion and action in 19-year-old caucasian men who had low birth weight. Diabetes. 2002 Apr;51(4):1271-80 PMID 11916955
- ↑ Park JH, Stoffers DA, Nicholls RD, Simmons RA. Development of type 2 diabetes following intrauterine growth retardation in rats is associated with progressive epigenetic silencing of Pdx1. J Clin Invest. 2008 Jun;118(6):2316-24 PMID 18464933
- 1 2 3 Campion J, Milagro FI, Martinez JA. Individuality and epigenetics in obesity. Obes Rev. 2009 Jul;10(4):383-92 doi:10.1111/j.1467-789X.2009.00595.x PMID 19413700
- ↑ Dunlevy LP, Burren KA, Mills K, Chitty LS, Copp AJ, Greene ND. Integrity of the methylation cycle is essential for mammalian neural tube closure. Birth Defects Res A Clin Mol Teratol. 2006 Jul;76(7):544-52 doi:10.1002/bdra.20286 PMID 16933307
- ↑ Linask KK, Huhta J. Folate protection from congenital heart defects linked with canonical wnt signaling and epigenetics. Curr Opin Pediatr. 2010 Oct;22(5):561-6 doi:10.1097/MOP.0b013e32833e2723 PMID 20844350 .
- ↑ Frye, R. E.; Slattery, J.; Delhey, L.; Furgerson, B.; Strickland, T.; Tippett, M.; Sailey, A.; Wynne, R.; Rose, S. (2016-10-18). "Folinic acid improves verbal communication in children with autism and language impairment: a randomized double-blind placebo-controlled trial". Molecular Psychiatry. ISSN 1476-5578. PMID 27752075. doi:10.1038/mp.2016.168.
- ↑ Jones G, Dwyer T. Birth weight, birth length, and bone density in prepubertal children: Evidence for an association that may be mediated by genetic factors. Calcif Tissue Int. 2000 Oct;67(4):304-8 PMID 11000344
- ↑ Javaid MK, Crozier SR, Harvey NC, Gale CR, Dennison EM, Boucher BJ, Arden NK, Godfrey KM, Cooper C, Princess Anne Hospital Study Group. Maternal vitamin D status during pregnancy and childhood bone mass at age 9 years: A longitudinal study. Lancet. 2006 Jan 7;367(9504):36-43 PMID 16399151
- ↑ Cooper C, Harvey N, Cole Z, Hanson M, Dennison E. Developmental origins of osteoporosis: The role of maternal nutrition. Adv Exp Med Biol. 2009;646:3h1-9 PMID 19536660
- ↑ Richards M, Hardy R, Kuh D, Wadsworth ME. Birth weight and cognitive function in the british 1946 birth cohort: Longitudinal population based study. BMJ. 2001 Jan 27;322(7280):199-203 PMID 11159613
- ↑ Gunnell D, Harrison G, Whitley E, Lewis G, Tynelius P, Rasmussen F. The association of fetal and childhood growth with risk of schizophrenia. cohort study of 720,000 swedish men and women. Schizophr Res. 2005 Nov 15;79(2-3):315-22 PMID 16125903.
- ↑ Lucas A. Programming by early nutrition: An experimental approach. J Nutr. 1998 Feb;128(2 Suppl):401S-6S PMID 9478036
- ↑ Mehedint MG, Niculescu MD, Craciunescu CN, Zeisel SH. Choline deficiency alters global histone methylation and epigenetic marking at the Re1 site of the calbindin 1 gene. FASEB J. 2010 Jan;24(1):184-95 PMID 19752176
- ↑ Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med. 1986 May 22;314(21):1360-8 doi:10.1056/NEJM198605223142106 PMID 3517648
- ↑ Ziegler AG, Schmid S, Huber D, Hummel M, Bonifacio E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA. 2003 Oct 1;290(13):1721-8 doi:10.1001/jama.290.13.1721 PMID 14519706
- 1 2 Davis CD, Ross SA. Dietary components impact histone modifications and cancer risk. Nutr Rev. 2007 Feb;65(2):88-94 PMID 17345961
- ↑ Martin JF, Johnston CR, Han CT, Benyshek DC. Nutritional origins of insulin resistance: a rat model for diabetes-prone human populations. 2000. J Nutr. 130:741-44 PMID 10736323.
- ↑ Zambrano E, Martinez-Samayoa PM, Bautista CJ, Deas M, Guillen L, Rodriguez-Gonzalez GL, Guzman C, Larrea F & Nathanielsz PW. Sex differences in transgenerational alterations of growth and metabolism in progeny (F2) of female offspring (F1) of rats fed a low protein diet during pregnancy and lactation. 2005. J Physiol (Lond.) 566:225-36 PMID 15860532
- 1 2 Harrison M & Langley-Evans SC. Intergenerational programming of impaired nephrogenesis and hypertension in rats following maternal protein restriction during pregnancy. 2009. Br. J. Nutr. 101: 1020-30 PMID 18778527
- 1 2 Benyshek DC, Johnston CS & Martin JF. Glucose metabolism is altered in the adequately-nourished grand-offspring (F3 generation) of rats malnourished during gestation and perinatal life. 2006. Diabetologia. 49: 1117-19 PMID 16557373
- ↑ Burdge GC, Slater-Jefferies J, Torrens C, Phillips ES, Hanson MA & Lillycrop KA. Dietary protein restriction of pregnant rats in the F0 generation induces altered methylation of hepatic gene promoters in the adult male offspring in the F1 and F2 generations. 2007. Br J Nutr 97(3): 435-9 doi:10.1017/S0007114507352392 PMID 17313703
- ↑ Boloker J, Gertz SJ & Simmons RA. Gestational diabetes leads to the development of diabetes in adulthood in the rat. 2002. Diabetes. 51(5): 1499-1506 PMID 11978648.
- 1 2 3 4 5 6 Fernandez-Twinn DS & Ozanne SE. Early life nutrition and metabolic programming. Ann N Y Acad Sci. Epub ahead of print 2010 Nov 11 PMID 21070247
- ↑ Niculescu, MD & Zeisel, SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. 2002. J Nutr. 132, 23335-23355 PMID 12163687
- ↑ Waterland, R. A. & Jirtle, R. L. Transposable elements: targets for early nutritional effects on epigenetic gene regulation. 2003. Mol. Cell. Biol. 23, 5293–5300 doi:10.1128/MCB.23.15.5293-5300.2003 PMID 12861015.
- ↑ Sziv KS, Ndlovu MN, Haegeman G & Vanden Berghe W . Nature or nurture: let food be your epigenetic medicine in chronic inflammatory disorders. 2010. Biochem. Pharmacol. 80(12):1816-32 doi:10.1016/j.bcp.2010.07.029 PMID 20688047
- ↑ Barnett M, Bermingham E, McNabb W, Bassett S, Armstrong K, Rounce J & Roy N. Investigating micronutrients and epigenetic mechanisms in relation to inflammatory bowel disease. 2010. Mutat Res. 690(1): 71-80 doi:10.1016/j.mrfmmm.2010.02.006 PMID 20188748
- ↑ Zeisel S. What Choline Metabolism Can Tell Us About the Underlying Mechanisms of Fetal Alcohol Spectrum Disorders. 2011 Mol Neurobiol 2011 Jan 25 epub doi:10.1007/s12035-011-8165-5 PMID 21259123
- 1 2 Ferguson-Smith, AC & Patti, ME. You are what your dad ate. Cell Metab. 2011. 13(2):115-7 doi:10.1016/j.cmet.2011.01.011 PMID 21284975
- ↑ Ng, S.F., Lin, R.C., Laybutt, D.R., Barres, R., Owens, J.A., and Morris, M.J. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. 2010. Nature 467, 963–966 doi:10.1038/nature09491 PMID 20962845