Methylmalonyl-CoA mutase






| methylmalonyl-CoA mutase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 5.4.99.2 | ||||||||
| CAS number | 9023-90-9 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / EGO | ||||||||
| |||||||||
Methylmalonyl-CoA mutase (MCM), mitochondrial, also known as methylmalonyl-CoA isomerase, is a protein that in humans is encoded by the MUT gene. This vitamin B12-dependent enzyme catalyzes the isomerization of methylmalonyl-CoA to succinyl-CoA in humans. Mutations in MUT gene may lead to various types of methylmalonic aciduria.[5]
MCM was first identified in rat liver and sheep kidney in 1955. In its latent form, it is 750 amino acids in length. Upon entry to the mitochondria, the 32 amino acid mitochondrial leader sequence at the N-terminus of the protein is cleaved, forming the fully processed monomer. The monomers then associate into homodimers, and bind AdoCbl (one for each monomer active site) to form the final, active holoenzyme form.[6]
Structure
Gene
The MUT gene lies on the chromosome location of 6p12.3 and consists of 13 exons, spanning over 35kb.[7]
Protein
The mature enzyme is a homodimer with the N-terminal CoA binding domain and the C- terminal cobalamin-binding domain.[8]
Function
Methylmalonyl-CoA mutase is expressed in high concentrations in the kidney, in intermediate concentrations in the heart, ovaries, brain, muscle, and liver, and in low concentrations in the spleen.[6] The enzyme can be found all throughout the central nervous system (CNS).[6] MCM resides in the mitochondria, where a number of substances, including the branched-chain amino acids isoleucine and valine, as well as methionine, threonine, thymine and odd-chain fatty acids, are metabolized via methylmalonate semialdehyde (MMlSA) or propionyl-CoA (Pr-CoA) to a common compound - methylmalonyl-CoA (MMl-CoA). MCM catalyzes the reversible isomerisation of l‐methylmalonyl‐CoA to succinyl‐CoA, requiring cobalamin (vitamin B12) in the form of adenosylcobalamin (AdoCbl) as a cofactor. As an important step in propionate catabolism, this reaction is required for the degradation of odd-chain fatty acids, the amino acids valine, isoleucine, methionine, and threonine, and cholesterol,[9] funneling metabolites from the breakdown of these amino acids into the tricarboxylic acid cycle.[10]
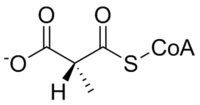
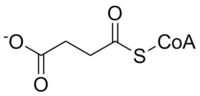
Methylmalonyl-CoA mutase catalyzes the following reaction:
| L-methylmalonyl-CoA | methylmalonyl-CoA mutase | Succinyl-CoA | |
 |
 | ||
| | |||
| methylmalonyl-CoA mutase | |||
The substrate of methylmalonyl-CoA mutase, methylmalonyl-CoA, is primarily derived from propionyl-CoA, a substance formed from the catabolism and digestion of isoleucine, valine, threonine, methionine, thymine, cholesterol, or odd-chain fatty acids.The product of the enzyme, succinyl-CoA, is a key molecule of the TCA cycle.
Clinical significance
A deficiency of this enzyme is responsible for an inherited disorder of metabolism, Methylmalonyl-CoA mutase deficiency, which is one of the causes of methylmalonic acidemia (also referred to as methylmalonic aciduria or MMA). MMA is an autosomal recessive inherited inborn error of metabolism, characterized by recurrent episodes of vomiting, lethargy, profound ketoacidosis, hyperammonemia, and pancytopenia in infancy, and may cause early death. Complications include cardiomyopathy, metabolic stroke, pancreatitis, and progressive renal failure.[5][11]
Either mutations to the gene MUT (encodes methylmalonyl-CoA mutase), or MMAA (encodes a chaperone protein of methylmalonyl-CoA mutase, MMAA protein) can lead to methylmalonyl acidemia.[12] Mutations to MUT can be categorized as either MUT0 (demonstrates no activity even in presence of excess AdoCbl), or MUT1 (demonstrates very low activity in presence of excess AdoCbl).[13] Over half of the mutations of MUT are missense mutations[14] while nonsense mutations comprise a significant remaining fraction (approximately 14%)[15]
Common treatment methods for MMA include a liver transplant or a liver and kidney transplant to combat the renal disease of methylmalonic acidemia. However, detrimental neurological effects can continue to plague patients even after a successful operation. It is thought that this is due to the widespread presence of methylmalonyl-CoA mutase throughout the central nervous system. Due to the loss of functionality of the enzyme, substrate levels build up in the CNS. The substrate, L-methylmalonyl-CoA hydrolyzes to form methylmalonate (methylmalonic acid), a neurotoxic dicarboxylic acid that, due to the poor dicarboxylic acid transport capacities of the blood-brain barrier, is effectively trapped within the CNS, leading to neurological debilitation. To combat these effects perioperative anti-catabolic regimes and no diet discontinuation are recommended.[6]
MUT has also been linked to bovine tuberculosis (bTB), a form of tuberculosis that affects mostly livestock and accounts for 10% of human cases.[16] It has been proposed that high MUT expression (thus high methylmalonyl-CoA mutase levels) leads to lower cholesterol levels which increases resistance to bTB and affords an improved response to the BCG vaccine.[16]
The murine model has proven an adequate and accurate way of studying the effects of MMA, and potential treatment methods.[17][18]
Mechanism
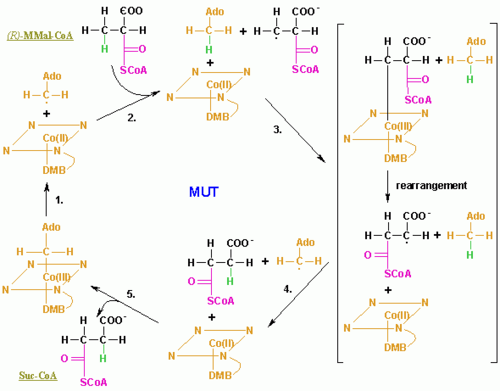
The MCM reaction mechanism begins with homolytic cleavage of AdoB12's C-Co(III) bond, the C and Co atoms each acquire one of the electrons that formed the cleaved electron pair bond. The Co ion, therefore, fluctuates between its Co(III) and Co(II) oxidation states [the two states are spectroscopically distinguishable: Co(III) is red and diamagnetic (no unpaired electrons), whereas Co(II) is yellow and paramagnetic (unpaired electrons)]. Hence, the role of coenzyme B-12 in the catalytic process is that of a reversible free radical generator. The C-Co(III) bond is well suited to this function because it is inherently weak (dissociation energy = 109 kJ/mol) and appears to be further weakened through steric interactions with the enzyme. A homolytic cleavage reaction is unusual in biology; most other biological bond cleavage reactions occur via heterolytic cleavage (in which the electron pair forming the cleaved bond is fully acquired by one of the separating atoms).
Methylmalonyl-CoA mutase is a member of the isomerase subfamily of adensylcobalamin-dependent enzymes. Furthermore, it is classified as class I, as it is a ‘DMB-off’/’His-on’ enzyme. This refers to the nature of the AdoCbl cofactor in the active site of methylmalonyl CoA.[19] AdoCbl is composed of a central cobalt-containing Corrin ring, an upper axial ligand (β-axial ligand), and a lower axial ligand (α-axial ligand). Both axial ligands are initially bonded to the central cobalt atom. In methylmalonyl-CoA mutase the β-axial ligand is 5’-deoxy-5’-adenosine and is involved in the free radical chemistry of the reaction. The α-axial ligand is 5,6-dimethylbenzimidazole (DMB) and is involved in organizing the active site to enable Histidine-610 to bond with Co, instead of DMB (the reason for the ‘DMB-off’/’His-on’ notation).[19] The Histidine-610 residue is critical to enzyme functionality. Its binding increases the rate of homolytic β-axial ligand – Co bond breakage by a factor of 1012.[20]

Other important residues of methylmalonyl-CoA mutase include Histidine-244, which acts as a general acid near the substrate and shields the radical species from side reactions involving oxygen,[23] Glutamate-370, whose hydrogen bond with the 2’-OH group of the ribose of the β-axial ligand forces interaction between the β-axial ligand radical species and the substrate,[24] and tyrosine-89 which stabilizes reactive radical intermediates and accounts for the stereo-selectivity of the enzyme.[21][25]
The processing protein, MMAA protein, fills the important role of aiding cofactor loading and exchange.[12][26] MMAA protein favors association with the MCM apoenzyme, and allows for the transfer of the AdoCbl cofactor to the enzyme active site.[26] Furthermore, if the bound AdoCbl accrues oxidative damage during normal functioning, MMAA protein fosters exchange of the damaged cofactor for a new AdoCbl via a GTP-reliant pathway.[12][26]
Interactions
References
- 1 2 3 GRCh38: Ensembl release 89: ENSG00000146085 - Ensembl, May 2017
- 1 2 3 GRCm38: Ensembl release 89: ENSMUSG00000023921 - Ensembl, May 2017
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- 1 2 Keyfi, F.; Sankian, M.; Moghaddassian, M.; Rolfs, A.; Varasteh, A. R. (2016-01-15). "Molecular, biochemical, and structural analysis of a novel mutation in patients with methylmalonyl-CoA mutase deficiency". Gene. 576 (1 Pt 2): 208–213. ISSN 1879-0038. PMID 26449400. doi:10.1016/j.gene.2015.10.002.
- 1 2 3 4 Ballhausen D, Mittaz L, Boulat O, Bonafé L, Braissant O (2009). "Evidence for catabolic pathway of propionate metabolism in CNS: expression pattern of methylmalonyl-CoA mutase and propionyl-CoA carboxylase alpha-subunit in developing and adult rat brain". Neuroscience. 164 (2): 578–87. PMID 19699272. doi:10.1016/j.neuroscience.2009.08.028.
- ↑ Chandler, R. J.; Venditti, C. P. (2016-10-01). "Genetic and genomic systems to study methylmalonic acidemia". Molecular Genetics and Metabolism. 86 (1–2): 34–43. ISSN 1096-7192. PMC 2657357
 . PMID 16182581. doi:10.1016/j.ymgme.2005.07.020.
. PMID 16182581. doi:10.1016/j.ymgme.2005.07.020. - ↑ Drennan, C. L.; Matthews, R. G.; Rosenblatt, D. S.; Ledley, F. D.; Fenton, W. A.; Ludwig, M. L. (1996-05-28). "Molecular basis for dysfunction of some mutant forms of methylmalonyl-CoA mutase: deductions from the structure of methionine synthase". Proceedings of the National Academy of Sciences of the United States of America. 93 (11): 5550–5555. ISSN 0027-8424. PMC 39284 . PMID 8643613. doi:10.1073/pnas.93.11.5550.
- ↑ Martínez, Maria Angeles; Rincón, Ana; Desviat, Lourdes R.; Merinero, Begoña; Ugarte, Magdalena; Pérez, Belén (2005-04-01). "Genetic analysis of three genes causing isolated methylmalonic acidemia: identification of 21 novel allelic variants". Molecular Genetics and Metabolism. 84 (4): 317–325. ISSN 1096-7192. PMID 15781192. doi:10.1016/j.ymgme.2004.11.011.
- ↑ Forny, Patrick; Froese, D. Sean; Suormala, Terttu; Yue, Wyatt W.; Baumgartner, Matthias R. (2014-12-01). "Functional characterization and categorization of missense mutations that cause methylmalonyl-CoA mutase (MUT) deficiency". Human Mutation. 35 (12): 1449–1458. ISSN 1098-1004. PMC 4441004 . PMID 25125334. doi:10.1002/humu.22633.
- ↑ Dündar, Halil; Özgül, Riza Köksal; Güzel-Ozantürk, Ayşegül; Dursun, Ali; Sivri, Serap; Aliefendioğlu, Didem; Coşkun, Turgay; Tokatli, Ayşegül (2012-08-01). "Microarray based mutational analysis of patients with methylmalonic acidemia: identification of 10 novel mutations". Molecular Genetics and Metabolism. 106 (4): 419–423. ISSN 1096-7206. PMID 22727635. doi:10.1016/j.ymgme.2012.05.014.
- 1 2 3 Takahashi-Íñiguez T, García-Arellano H, Trujillo-Roldán MA, Flores ME (2011). "Protection and reactivation of human methylmalonyl-CoA mutase by MMAA protein". Biochem. Biophys. Res. Commun. 404 (1): 443–7. PMID 21138732. doi:10.1016/j.bbrc.2010.11.141.
- ↑ Drennan CL, Matthews RG, Rosenblatt DS, Ledley FD, Fenton WA, Ludwig ML (1996). "Molecular basis for dysfunction of some mutant forms of methylmalonyl-CoA mutase: deductions from the structure of methionine synthase". Proc. Natl. Acad. Sci. U.S.A. 93 (11): 5550–5. PMC 39284 . PMID 8643613. doi:10.1073/pnas.93.11.5550.
- ↑ Forny P, Froese DS, Suormala T, Yue WW, Baumgartner MR (2014). "Functional characterization and categorization of missense mutations that cause methylmalonyl-CoA mutase (MUT) deficiency". Hum. Mutat. 35 (12): 1449–58. PMC 4441004 . PMID 25125334. doi:10.1002/humu.22633.
- ↑ Buck NE, Wood LR, Hamilton NJ, Bennett MJ, Peters HL (2012). "Treatment of a methylmalonyl-CoA mutase stopcodon mutation". Biochem. Biophys. Res. Commun. 427 (4): 753–7. PMID 23041189. doi:10.1016/j.bbrc.2012.09.133.
- 1 2 de la Fuente J, Gortazar C, Vicente J, Villar M (2011). "Host expression of methylmalonyl-CoA mutase and tuberculosis: a missing link?". Med. Hypotheses. 76 (3): 361–4. PMID 21084167. doi:10.1016/j.mehy.2010.10.040.
- ↑ Chandler RJ, Venditti CP (2012). "Pre-clinical efficacy and dosing of an AAV8 vector expressing human methylmalonyl-CoA mutase in a murine model of methylmalonic acidemia (MMA)". Mol. Genet. Metab. 107 (3): 617–9. PMC 3522145 . PMID 23046887. doi:10.1016/j.ymgme.2012.09.019.
- ↑ Manoli I, Sysol JR, Li L, Houillier P, Garone C, Wang C, Zerfas PM, Cusmano-Ozog K, Young S, Trivedi NS, Cheng J, Sloan JL, Chandler RJ, Abu-Asab M, Tsokos M, Elkahloun AG, Rosen S, Enns GM, Berry GT, Hoffmann V, DiMauro S, Schnermann J, Venditti CP (2013). "Targeting proximal tubule mitochondrial dysfunction attenuates the renal disease of methylmalonic acidemia". Proc. Natl. Acad. Sci. U.S.A. 110 (33): 13552–7. PMC 3746875 . PMID 23898205. doi:10.1073/pnas.1302764110.
- 1 2 Takahashi-Iñiguez T, García-Hernandez E, Arreguín-Espinosa R, Flores ME (2012). "Role of vitamin B12 on methylmalonyl-CoA mutase activity". J Zhejiang Univ Sci B. 13 (6): 423–37. PMC 3370288 . PMID 22661206. doi:10.1631/jzus.B1100329.
- ↑ Vlasie M, Chowdhury S, Banerjee R (2002). "Importance of the histidine ligand to coenzyme B12 in the reaction catalyzed by methylmalonyl-CoA mutase". J. Biol. Chem. 277 (21): 18523–7. PMID 11893736. doi:10.1074/jbc.M111809200.
- 1 2 Mancia F, Smith GA, Evans PR (1999). "Crystal structure of substrate complexes of methylmalonyl-CoA mutase". Biochemistry. 38 (25): 7999–8005. PMID 10387043. doi:10.1021/bi9903852.
- ↑ Mancia F, Evans PR (1998). "Conformational changes on substrate binding to methylmalonyl CoA mutase and new insights into the free radical mechanism". Structure. 6 (6): 711–20. PMID 9655823. doi:10.1016/s0969-2126(98)00073-2.
- ↑ Maiti N, Widjaja L, Banerjee R (1999). "Proton transfer from histidine 244 may facilitate the 1,2 rearrangement reaction in coenzyme B(12)-dependent methylmalonyl-CoA mutase". J. Biol. Chem. 274 (46): 32733–7. PMID 10551831. doi:10.1074/jbc.274.46.32733.
- ↑ Buckel W, Friedrich P, Golding BT (2012). "Hydrogen bonds guide the short-lived 5'-deoxyadenosyl radical to the place of action". Angew. Chem. Int. Ed. Engl. 51 (40): 9974–6. PMID 22945861. doi:10.1002/anie.201205299.
- ↑ Thomä NH, Meier TW, Evans PR, Leadlay PF (1998). "Stabilization of radical intermediates by an active-site tyrosine residue in methylmalonyl-CoA mutase". Biochemistry. 37 (41): 14386–93. PMID 9772164. doi:10.1021/bi981375o.
- 1 2 3 Froese DS, Kochan G, Muniz JR, Wu X, Gileadi C, Ugochukwu E, Krysztofinska E, Gravel RA, Oppermann U, Yue WW (2010). "Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation". J. Biol. Chem. 285 (49): 38204–13. PMC 2992254 . PMID 20876572. doi:10.1074/jbc.M110.177717.
- 1 2 Froese, D. Sean; Kochan, Grazyna; Muniz, João R. C.; Wu, Xuchu; Gileadi, Carina; Ugochukwu, Emelie; Krysztofinska, Ewelina; Gravel, Roy A.; Oppermann, Udo (2010-12-03). "Structures of the human GTPase MMAA and vitamin B12-dependent methylmalonyl-CoA mutase and insight into their complex formation". The Journal of Biological Chemistry. 285 (49): 38204–38213. ISSN 1083-351X. PMC 2992254 . PMID 20876572. doi:10.1074/jbc.M110.177717.
- ↑ Padovani, Dominique; Labunska, Tetyana; Banerjee, Ruma (2006-06-30). "Energetics of interaction between the G-protein chaperone, MeaB, and B12-dependent methylmalonyl-CoA mutase". The Journal of Biological Chemistry. 281 (26): 17838–17844. ISSN 0021-9258. PMID 16641088. doi:10.1074/jbc.M600047200.
- ↑ Taoka, S.; Padmakumar, R.; Lai, M. T.; Liu, H. W.; Banerjee, R. (1994-12-16). "Inhibition of the human methylmalonyl-CoA mutase by various CoA-esters". The Journal of Biological Chemistry. 269 (50): 31630–31634. ISSN 0021-9258. PMID 7989334.
Further reading
- Ledley FD, Rosenblatt DS (1997). "Mutations in mut methylmalonic acidemia: clinical and enzymatic correlations.". Hum. Mutat. 9 (1): 1–6. PMID 8990001. doi:10.1002/(SICI)1098-1004(1997)9:1<1::AID-HUMU1>3.0.CO;2-E.
- Ludwig ML, Matthews RG (1997). "Structure-based perspectives on B12-dependent enzymes.". Annu. Rev. Biochem. 66: 269–313. PMID 9242908. doi:10.1146/annurev.biochem.66.1.269.
- Lubrano R, Elli M, Rossi M, et al. (2007). "Renal transplant in methylmalonic acidemia: could it be the best option? Report on a case at 10 years and review of the literature.". Pediatr. Nephrol. 22 (8): 1209–14. PMID 17401587. doi:10.1007/s00467-007-0460-z.
- Frenkel EP, Kitchens RL (1978). "Intracellular localization of hepatic propionyl-CoA carboxylase and methylmalonyl-CoA mutase in humans and normal and vitamin B12 deficient rats.". Br. J. Haematol. 31 (4): 501–13. PMID 24458. doi:10.1111/j.1365-2141.1975.tb00885.x.
- Crane AM, Jansen R, Andrews ER, Ledley FD (1992). "Cloning and expression of a mutant methylmalonyl coenzyme A mutase with altered cobalamin affinity that causes mut- methylmalonic aciduria.". J. Clin. Invest. 89 (2): 385–91. PMC 442864 . PMID 1346616. doi:10.1172/JCI115597.
- Crane AM, Martin LS, Valle D, Ledley FD (1992). "Phenotype of disease in three patients with identical mutations in methylmalonyl CoA mutase.". Hum. Genet. 89 (3): 259–64. PMID 1351030. doi:10.1007/BF00220536.
- Raff ML, Crane AM, Jansen R, et al. (1991). "Genetic characterization of a MUT locus mutation discriminating heterogeneity in mut0 and mut- methylmalonic aciduria by interallelic complementation.". J. Clin. Invest. 87 (1): 203–7. PMC 295026 . PMID 1670635. doi:10.1172/JCI114972.
- Jansen R, Ledley FD (1990). "Heterozygous mutations at the mut locus in fibroblasts with mut0 methylmalonic acidemia identified by polymerase-chain-reaction cDNA cloning.". Am. J. Hum. Genet. 47 (5): 808–14. PMC 1683687 . PMID 1977311.
- Nham SU, Wilkemeyer MF, Ledley FD (1991). "Structure of the human methylmalonyl-CoA mutase (MUT) locus.". Genomics. 8 (4): 710–6. PMID 1980486. doi:10.1016/0888-7543(90)90259-W.
- Ledley FD, Lumetta M, Nguyen PN, et al. (1988). "Molecular cloning of L-methylmalonyl-CoA mutase: gene transfer and analysis of mut cell lines.". Proc. Natl. Acad. Sci. U.S.A. 85 (10): 3518–21. PMC 280243 . PMID 2453061. doi:10.1073/pnas.85.10.3518.
- Jansen R, Kalousek F, Fenton WA, et al. (1989). "Cloning of full-length methylmalonyl-CoA mutase from a cDNA library using the polymerase chain reaction.". Genomics. 4 (2): 198–205. PMID 2567699. doi:10.1016/0888-7543(89)90300-5.
- Fenton WA, Hack AM, Kraus JP, Rosenberg LE (1987). "Immunochemical studies of fibroblasts from patients with methylmalonyl-CoA mutase apoenzyme deficiency: detection of a mutation interfering with mitochondrial import.". Proc. Natl. Acad. Sci. U.S.A. 84 (5): 1421–4. PMC 304442 . PMID 2881300. doi:10.1073/pnas.84.5.1421.
- Zoghbi HY, O'Brien WE, Ledley FD (1989). "Linkage relationships of the human methylmalonyl CoA mutase to the HLA and D6S4 loci on chromosome 6.". Genomics. 3 (4): 396–8. PMID 2907507. doi:10.1016/0888-7543(88)90135-8.
- Kolhouse JF, Utley C, Allen RH (1980). "Isolation and characterization of methylmalonyl-CoA mutase from human placenta.". J. Biol. Chem. 255 (7): 2708–12. PMID 6102092.
- Fenton WA, Hack AM, Willard HF, et al. (1982). "Purification and properties of methylmalonyl coenzyme A mutase from human liver.". Arch. Biochem. Biophys. 214 (2): 815–23. PMID 6124211. doi:10.1016/0003-9861(82)90088-1.
- Qureshi AA, Crane AM, Matiaszuk NV, et al. (1994). "Cloning and expression of mutations demonstrating intragenic complementation in mut0 methylmalonic aciduria.". J. Clin. Invest. 93 (4): 1812–9. PMC 294249 . PMID 7909321. doi:10.1172/JCI117166.
- Crane AM, Ledley FD (1994). "Clustering of mutations in methylmalonyl CoA mutase associated with mut- methylmalonic acidemia.". Am. J. Hum. Genet. 55 (1): 42–50. PMC 1918235 . PMID 7912889.
- Janata J, Kogekar N, Fenton WA (1998). "Expression and kinetic characterization of methylmalonyl-CoA mutase from patients with the mut- phenotype: evidence for naturally occurring interallelic complementation.". Hum. Mol. Genet. 6 (9): 1457–64. PMID 9285782. doi:10.1093/hmg/6.9.1457.
External links
- GeneReviews/NIH/NCBI/UW entry on Methylmalonic Acidemia
- Methylmalonyl-CoA Mutase at the US National Library of Medicine Medical Subject Headings (MeSH)