Metalloprotein

Metalloprotein is a generic term for a protein that contains a metal ion cofactor.[1][2] A large number of all proteins are part of this category.
Function
It is estimated that approximately half of all proteins contain a metal.[3] In another estimate, about one quarter to one third of all proteins are proposed to require metals to carry out their functions.[4] Thus, metalloproteins have many different functions in cells, such as storage and transport of proteins, enzymes and signal transduction proteins. The role of metal ions in infectious diseases has been reviewed.[5]
Coordination chemistry principles
In metalloproteins, metal ions are usually coordinated by nitrogen, oxygen or sulfur centers belonging to amino acid residues of the protein. These donor groups are often provided by side-chains on the amino acid residues. Especially important are the imidazole substituent in histidine residues, thiolate substituents in cysteine residues, and carboxylate groups provided by aspartate. Given the diversity of the metalloproteome, virtually all amino acid residues have been shown to bind metal centers. The peptide backbone also provides donor groups; these include deprotonated amides and the amide carbonyl oxygen centers. Lead(II) binding in natural and artificial proteins has been reviewed.[6]
In addition to donor groups that are provided by amino acid residues, a large number of organic cofactors function as ligands. Perhaps most famous are the tetradentate N4 macrocyclic ligands incorporated into the heme protein. Inorganic ligands such as sulfide and oxide are also common.
Storage and transport metalloproteins
These are the second stage product of protein hydrolysis obtained by treatment with slightly stronger acids and alkalies
Oxygen carriers
Hemoglobin, which is the principal oxygen-carrier in humans, has four subunits in which the iron(II) ion is coordinated by the planar macrocyclic ligand protoporphyrin IX (PIX) and the imidazole nitrogen atom of a histidine residue. The sixth coordination site contains a water molecule or a dioxygen molecule. By contrast the protein myoglobin, found in muscle cells, has only one such unit. The active site is located in an hydrophobic pocket. This is important as without it the iron(II) would be irreversibly oxidized to iron(III). The equilibrium constant for the formation of HbO2 is such that oxygen is taken up or released depending on the partial pressure of oxygen in the lungs or in muscle. In hemoglobin the four subunits show a cooperativity effect that allows for easy oxygen transfer from hemoglobin to myoglobin.[7]
In both hemoglobin and myoglobin it is sometimes incorrectly stated that the oxygenated species contains iron(III). It is now known that the diamagnetic nature of these species is because the iron(II) atom is in the low-spin state. In oxyhemoglobin the iron atom is located in the plane of the porphyrin ring, but in the paramagnetic deoxyhemoglobin the iron atom lies above the plane of the ring.[7] This change in spin state is a cooperative effect due to the higher crystal field splitting and smaller ionic radius of Fe2+ in the oxyhemoglobin moiety.
Hemerythrin is another iron-containing oxygen carrier. The oxygen binding site is a binuclear iron center. The iron atoms are coordinated to the protein through the carboxylate side chains of a glutamate and aspartate and five histidine residues. The uptake of O2 by hemerythrin is accompanied by two-electron oxidation of the reduced binuclear center to produce bound peroxide (OOH−). The mechanism of oxygen uptake and release have been worked out in detail.[8][9]
Hemocyanins carry oxygen in the blood of most mollusks, and some arthropods such as the horseshoe crab. They are second only to hemoglobin in biological popularity of use in oxygen transport. On oxygenation the two copper(I) atoms at the active site are oxidized to copper(II) and the dioxygen molecules are reduced to peroxide, O2−
2.[10][11]
Chlorocruorin (as the larger carrier erythrocruorin) is an oxygen-binding hemeprotein present in the blood plasma of many annelids, particularly certain marine polychaetes.
Cytochromes
Oxidation and reduction reactions are not common in organic chemistry as few organic molecules can act as oxidizing or reducing agents. Iron(II), on the other hand, can easily be oxidized to iron(III). This functionality is used in cytochromes, which function as electron-transfer vectors. The presence of the metal ion allows metalloenzymes to perform functions such as redox reactions that cannot easily be performed by the limited set of functional groups found in amino acids.[12] The iron atom in most cytochromes is contained in a heme group. The differences between those cytochromes lies in the different side-chains. For instance cytochrome a has a heme a prosthetic group and cytochrome b has a heme b prosthetic group. These differences result in different Fe2+/Fe3+ redox potentials such that various cytochromes are involved in the mitochondrial electron transport chain.[13]
Cytochrome P450 enzymes perform the function of inserting an oxygen atom into a C−H bond, an oxidation reaction.[14][15]

Rubredoxin
Rubredoxin is an electron-carrier found in sulfur-metabolizing bacteria and archaea. The active site contains an iron ion coordinated by the sulfur atoms of four cysteine residues forming an almost regular tetrahedron. Rubredoxins perform one-electron transfer processes. The oxidation state of the iron atom changes between the +2 and +3 states. In both oxidation states the metal is high spin, which helps to minimize structural changes.
Plastocyanin
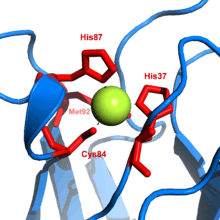
Plastocyanin is one of the family of blue copper proteins that are involved in electron transfer reactions. The copper-binding site is described as distorted trigonal pyramidal.[16] The trigonal plane of the pyramidal base is composed of two nitrogen atoms (N1 and N2) from separate histidines and a sulfur (S1) from a cysteine. Sulfur (S2) from an axial methionine forms the apex. The distortion occurs in the bond lengths between the copper and sulfur ligands. The Cu−S1 contact is shorter (207 pm) than Cu−S2 (282 pm). The elongated Cu−S2 bonding destabilizes the Cu(II) form and increases the redox potential of the protein. The blue color (597 nm peak absorption) is due to the Cu−S1 bond where S(pπ) to Cu(dx2−y2) charge transfer occurs.[17]
In the reduced form of plastocyanin, His-87 will become protonated with a pKa of 4.4. Protonation prevents it acting as a ligand and the copper site geometry becomes trigonal planar.
Metal-ion storage and transfer
Iron
Iron is stored as iron(III) in ferritin. The exact nature of the binding site has not yet been determined. The iron appears to be present as a hydrolysis product such as FeO(OH). Iron is transported by transferrin whose binding site consists of two tyrosines, one aspartic acid and one histidine.[18] The human body has no mechanism for iron excretion. This can lead to iron overload problems in patients treated with blood transfusions, as, for instance, with β-thalassemia. Iron is actually excreted in urine[19] and is also concentrated in bile[20] which is excreted in feces.[21]
Copper
Ceruloplasmin is the major copper-carrying protein in the blood. Ceruloplasmin exhibits oxidase activity, which is associated with possible oxidation of Fe(II) into Fe(III), therefore assisting in its transport in the blood plasma in association with transferrin, which can carry iron only in the Fe(III) state.
Calcium
Osteopontin is involved in mineralization in the extracellular matrices of bones and teeth.
Metalloenzymes
Metalloenzymes all have one feature in common, namely that the metal ion is bound to the protein with one labile coordination site. As with all enzymes, the shape of the active site is crucial. The metal ion is usually located in a pocket whose shape fits the substrate. The metal ion catalyzes reactions that are difficult to achieve in organic chemistry.
Carbonic anhydrase
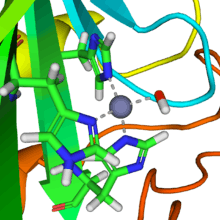
In aqueous solution, carbon dioxide forms carbonic acid
- CO2 + H2O ⇌ H2CO3
This reaction is very slow in the absence of a catalyst, but quite fast in the presence of the hydroxide ion
- CO2 + OH− ⇌ HCO−
3
A reaction similar to this is almost instantaneous with carbonic anhydrase. The structure of the active site in carbonic anhydrases is well-known from a number of crystal structures. It consists of a zinc ion coordinated by three imidazole nitrogen atoms from three histidine units. The fourth coordination site is occupied by a water molecule. The coordination sphere of the zinc ion is approximately tetrahedral. The positively-charged zinc ion polarizes the coordinated water molecule, and nucleophilic attack by the negatively-charged hydroxide portion on carbon dioxide (carbonic anhydride) proceeds rapidly. The catalytic cycle produces the bicarbonate ion and the hydrogen ion[2] as the equilibrium
- H2CO3 ⇌ HCO−
3 + H+
favours dissociation of carbonic acid at biological pH values.[22]
Vitamin B12-dependent enzymes
The cobalt-containing Vitamin B12 (also known as cobalamin) catalyzes the transfer of methyl (−CH3) groups between two molecules, which involves the breaking of C−C bonds, a process that is energetically expensive in organic reactions. The metal ion lowers the activation energy for the process by forming a transient Co−CH3 bond.[23] The structure of the coenzyme was famously determined by Dorothy Hodgkin and co-workers, for which she received a Nobel Prize in Chemistry.[24] It consists of a cobalt(II) ion coordinated to four nitrogen atoms of a corrin ring and a fifth nitrogen atom from an imidazole group. In the resting state there is a Co−C sigma bond with the 5′ carbon atom of adenosine.[25] This is a naturally occurring organometallic compound, which explains its function in trans-methylation reactions, such as the reaction carried out by methionine synthase.
Nitrogenase (nitrogen fixation)
The fixation of atmospheric nitrogen is a very energy-intensive process, as it involves breaking the very stable triple bond between the nitrogen atoms. The enzyme nitrogenase is one of the few enzymes that can catalyze the process. The enzyme occurs in Rhizobium bacteria. There are three components to its action: a molybdenum atom at the active site, iron–sulfur clusters that are involved in transporting the electrons needed to reduce the nitrogen, and an abundant energy source in the form of magnesium ATP. This last is provided by a symbiotic relationship between the bacteria and a host plant, often a legume. The relationship is symbiotic because the plant supplies the energy by photosynthesis and benefits by obtaining the fixed nitrogen. The reaction may be written symbolically as
where Pi stands for inorganic phosphate. The precise structure of the active site has been difficult to determine. It appears to contain a MoFe7S8 cluster that is able to bind the dinitrogen molecule and, presumably, enable the reduction process to begin.[26] The electrons are transported by the associated "P" cluster, which contains two cubical Fe4S4 clusters joined by sulfur bridges.[27]
Superoxide dismutase

The superoxide ion, O−
2 is generated in biological systems by reduction of molecular oxygen. It has an unpaired electron, so it behaves as a free radical. It is a powerful oxidizing agent. These properties render the superoxide ion very toxic and are deployed to advantage by phagocytes to kill invading microorganisms. Otherwise, the superoxide ion must be destroyed before it does unwanted damage in a cell. The superoxide dismutase enzymes perform this function very efficiently.[28]
The formal oxidation state of the oxygen atoms is − 1⁄2. In solutions at neutral pH, the superoxide ion disproportionates to molecular oxygen and hydrogen peroxide.
- 2 O−
2 + 2 H+ → O2 + H2O2
In biology this type of reaction is called a dismutation reaction. It involves both oxidation and reduction of superoxide ions. The superoxide dismutase (SOD) group of enzymes increase the rate of reaction to near the diffusion-limited rate.[29] The key to the action of these enzymes is a metal ion with variable oxidation state that can act either as an oxidizing agent or as a reducing agent.
- Oxidation: M(n+1)+ + O−
2 → Mn+ + O2 - Reduction: Mn+ + O−
2 + 2 H+ → M(n+1)+ + H2O2.
In human SOD the active metal is copper, as Cu(II) or Cu(I), coordinated tetrahedrally by four histidine residues. This enzyme also contains zinc ions for stabilization and is activated by copper chaperone for superoxide dismutase (CCS). Other isozymes may contain iron, manganese or nickel. Ni-SOD is particularly interesting as it involves nickel(III), an unusual oxidation state for this element. The active site nickel geometry cycles from square planar Ni(II), with thiolate (Cys2 and Cys6) and backbone nitrogen (His1 and Cys2) ligands, to square pyramidal Ni(III) with an added axial His1 side chain ligand.[30]
Chlorophyll-containing proteins

Chlorophyll plays a crucial role in photosynthesis. It contains a magnesium enclosed in a chlorin ring. However, the magnesium ion is not directly involved in the photosynthetic function and can be replaced by other divalent ions with little loss of activity. Rather, the photon is absorbed by the chlorin ring, whose electronic structure is well-adapted for this purpose.
Initially, the absorption of a photon causes an electron to be excited into a singlet state of the Q band. The excited state undergoes an intersystem crossing from the singlet state to a triplet state in which there are two electrons with parallel spin. This species is, in effect, a free radical, and is very reactive and allows an electron to be transferred to acceptors that are adjacent to the chlorophyll in the chloroplast. In the process chlorophyll is oxidized. Later in the photosynthetic cycle, chlorophyll is reduced back again. This reduction ultimately draws electrons from water, yielding molecular oxygen as a final oxidation product.
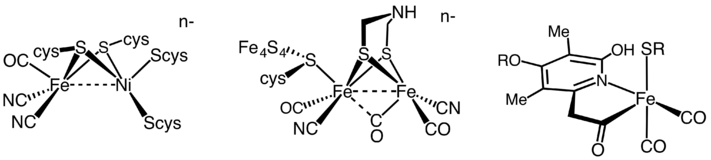
Hydrogenase
Hydrogenases are subclassified into three different types based on the active site metal content: iron–iron hydrogenase, nickel–iron hydrogenase, and iron hydrogenase.[31] All hydrogenases catalyze reversible H2 uptake, but while the [FeFe] and [NiFe] hydrogenases are true redox catalysts, driving H2 oxidation and H+ reduction
- H2 ⇌ 2 H+ + 2 e−
the [Fe] hydrogenases catalyze the reversible heterolytic cleavage of H2.
- H2 ⇌ H+ + H−
Ribozyme and deoxyribozyme
Since discovery of ribozymes by Thomas Cech and Sidney Altman in the early 1980s, ribozymes has been shown to be a distinct class of metalloenzymes.[32] Many ribozymes require metal ions in their active sites for chemical catalysis; hence they are called metalloenzymes. Additionally, metal ions are essential for the stabilization of ribozyme structure. Group I intron is the most studied ribozyme which has three metals participating in catalysis.[33] Other known ribozymes include group II intron, RNase P, and several small viral ribozymes (such as hammerhead, hairpin, HDV, and VS). Recently, four new classes of ribozymes have been discovered (named twister, twister sister, pistol and hatchet) which are all self-cleaving ribozymes.[34]
Deoxyribozymes, also called DNAzymes or catalytic DNA, are first discovered in 1994 and quickly emerged as a new class of metalloenzymes.[35] Almost all DNAzymes require metal ions for their function; thus they are classified as metalloenzymes. Although ribozymes mostly catalyze cleavage of RNA substrates, variety of reactions can be catalyzed by DNAzymes including RNA/DNA cleavage, RNA/DNA ligation, amino acid phosphorylation and dephosphorylation, and carbon–carbon bond formation.[36] Yet, DNAzymes that catalyze RNA cleavage reaction are the most extensively explored ones. 10-23 DNAzyme, discovered in 1997, is one of the most studied catalytic DNAs with clinical applications as a therapeutic agent.[37] Several metal-specific DNAzymes have been reported including the GR-5 DNAzyme (lead-specific),[38] the CA1-3 DNAzymes (copper-specific), the 39E DNAzyme (uranyl-specific)[39] and the NaA43 DNAzyme (sodium-specific).[40]
Signal-transduction metalloproteins
Calmodulin

Calmodulin is an example of a signal-transduction protein. It is a small protein that contains four EF-hand motifs, each of which able to bind a Ca2+ ion.
In an EF-hand loop the calcium ion is coordinated in a pentagonal bipyramidal configuration. Six glutamic acid and aspartic acid residues involved in the binding are in positions 1, 3, 5, 7 and 9 of the polypeptide chain. At position 12, there is a glutamate or aspartate ligand that behaves as a (bidentate ligand), providing two oxygen atoms. The ninth residue in the loop is necessarily glycine due to the conformational requirements of the backbone. The coordination sphere of the calcium ion contains only carboxylate oxygen atoms and no nitrogen atoms. This is consistent with the hard nature of the calcium ion.
The protein has two approximately symmetrical domains, separated by a flexible "hinge" region. Binding of calcium causes a conformational change to occur in the protein. Calmodulin participates in an intracellular signaling system by acting as a diffusible second messenger to the initial stimuli.[41][42]
Troponin
In both cardiac and skeletal muscles, muscular force production is controlled primarily by changes in the intracellular calcium concentration. In general, when calcium rises, the muscles contract and, when calcium falls, the muscles relax. Troponin, along with actin and tropomyosin, is the protein complex to which calcium binds to trigger the production of muscular force.
Transcription factors
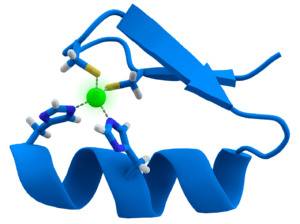
Many transcription factors contain a structure known as a zinc finger, this is a structural module where a region of protein folds around a zinc ion. The zinc does not directly contact the DNA that these proteins bind to. Instead, the cofactor is essential for the stability of the tightly-folded protein chain.[43] In these proteins, the zinc ion is usually coordinated by pairs of cysteine and histidine side-chains.
Other metalloenzymes
There are two types of carbon monoxide dehydrogenase: one contains copper and molybdenum, the other contains nickel and iron. Parallels and differences in catalytic strategies have been reviewed.[44]
Pb2+ (lead) can replace Ca2+ (calcium) as, for example, with calmodulin or Zn2+ (zinc) as with metallocarboxypeptidases[45]
Some other metalloenzymes are given in the following table, according to the metal involved.
See also
References
- ↑ Banci, Lucia (2013). Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O., eds. Metallomics and the Cell. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402.
- 1 2 Shriver, D. F.; Atkins, P. W. (1999). "Chapter 19, Bioinorganic chemistry". Inorganic chemistry (3rd ed.). Oxford University Press. ISBN 0-19-850330-X.
- ↑ Thomson, A. J.; Gray, H. B. (1998). "Bioinorganic chemistry". Current Opinion in Chemical Biology. 2: 155–158. doi:10.1016/S1367-5931(98)80056-2.
- ↑ Waldron, K. J.; Robinson, N. J. (January 2009). "How do bacterial cells ensure that metalloproteins get the correct metal?". Nat. Rev. Microbiol. 7 (1): 25–35. PMID 19079350. doi:10.1038/nrmicro2057.
- ↑ Carver, Peggy L. (2013). "Chapter 1. Metal Ions and Infectious Diseases. An Overview from the Clinic". In Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O. Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. 13. Springer. pp. 1–28. doi:10.1007/978-94-007-7500-8_1.
- ↑ Cangelosi, Virginia; Ruckthong, Leela; Pecoraro, Vincent L. (2017). "Chapter 10. Lead(II) Binding in Natural and Artificial Proteins". In Astrid, S.; Helmut, S.; Sigel, R. K. O. Lead: Its Effects on Environment and Health. Metal Ions in Life Sciences. 17. de Gruyter. pp. 271–318. doi:10.1515/9783110434330-010.
- 1 2 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9. Fig.25.7, p 1100 illustrates the structure of deoxyhemoglobin
- ↑ Stenkamp, R. E. (1994). "Dioxygen and hemerythrin". Chem. Rev. 94: 715–726. doi:10.1021/cr00027a008.
- ↑ Wirstam, M.; Lippard, S. J.; Friesner, R. A. (2003). "Reversioble Dioxygen Binding to Hemerythrin". J. Am. Chem. Soc. 125 (13): 3980–3987. PMID 12656634. doi:10.1021/ja017692r.
- ↑ Karlin, K. D.; Cruse, R. W.; Gultneh, Y.; Farooq, A.; Hayes, J. C.; Zubieta, J. (1987). "Dioxygen–copper reactivity. Reversible binding of O2 and CO to a phenoxo-bridged dicopper(I) complex". J. Am. Chem. Soc. 109 (9): 2668–2679. doi:10.1021/ja00243a019.
- ↑ Kitajima, N.; Fujisawa, K.; Fujimoto, C.; Morooka, Y.; Hashimoto, S.; Kitagawa, T.; Toriumi, K.; Tatsumi, K.; Nakamura, A. (1992). "A new model for dioxygen binding in hemocyanin. Synthesis, characterization, and molecular structure of the μ-η2:η2-peroxo dinuclear copper(II) complexes, [Cu(Hb(3,5-R2pz)3)]2(O2) (R = isopropyl and Ph)". J. Am. Chem. Soc. 114 (4): 1277–1291. doi:10.1021/ja00030a025.
- ↑ Messerschmidt, A; Huber, R.; Wieghardt, K.; Poulos, T. (2001). Handbook of Metalloproteins. Wiley. ISBN 0-471-62743-7.
- ↑ Moore, G. R.; Pettigrew,, G. W. (1990). Cytochrome c: Structural and Physicochemical Aspects. Berlin: Springer.
- ↑ Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O., eds. (2007). The Ubiquitous Roles of Cytochrome 450 Proteins. Metal Ions in Life Sciences. 3. Wiley. ISBN 978-0-470-01672-5.
- ↑ Ortiz de Montellano, P. R. (2005). Cytochrome P450 Structure, Mechanism, and Biochemistry (3rd ed.). Springer. ISBN 978-0-306-48324-0.
- ↑ Colman, P. M.; Freeman, H. C.; Guss, J. M.; Murata, M.; Norris, V. A.; Ramshaw, J. A. M.; Venkatappa, M. P. (1978). "X-Ray Crystal-Structure Analysis of Plastocyanin at 2.7 Å Resolution". Nature. 272 (5651): 319–324. doi:10.1038/272319a0.
- ↑ Solomon, E. I.; Gewirth, A. A.; Cohen, S. L. (1986). "Spectroscopic Studies of Active Sites. Blue Copper and Electronic Structural Analogs". ACS Symposium Series. 307: 236–266. doi:10.1021/bk-1986-0307.ch016.
- ↑ Anderson, B. F.; Baker, H. M.; Dodson, E. J.; et al. (April 1987). "Structure of human lactoferrin at 3.2 Å resolution". Proc. Natl. Acad. Sci. USA. 84 (7): 1769–73. PMC 304522
 . PMID 3470756. doi:10.1073/pnas.84.7.1769.
. PMID 3470756. doi:10.1073/pnas.84.7.1769. - ↑ "Iron, copper and zinc levels in urine".
- ↑ "Iron absorption and biliary excretion of transferrin in rats.".
- ↑ "Biliary excretion of waste products".
- ↑ Lindskog, S. (1997). "Structure and mechanism of carbonic anhydrase". Pharmacol. Ther. 74 (1): 1–20. PMID 9336012. doi:10.1016/S0163-7258(96)00198-2.
- ↑ Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O., eds. (2008). Metal–carbon bonds in enzymes and cofactors. Metal Ions in Life Sciences. 6. Wiley. ISBN 978-1-84755-915-9.
- ↑ "The Nobel Prize in Chemistry 1964". Nobelprize.org. Retrieved 2008-10-06.
- ↑ Hodgkin, D. C. (1965). "The Structure of the Corrin Nucleus from X-ray Analysis". Proc. Roy. Soc. A. 288: 294–305. doi:10.1098/rspa.1965.0219.
- ↑ Orme-Johnson, W. H. (1993). Steifel, E. I.; Coucouvannis, D.; Newton, D. C., eds. Molybdenum enzymes, cofactors and model systems. Advances in chemystry, Symposium series no. 535. Washington, DC: American Chemical Society. p. 257.
- ↑ Chan, M. K.; Kim, J.; Rees, D. C. (1993). "The nitrogenase FeMo-cofactor and P-cluster pair: 2.2 Å resolution structures". Science. 260 (5109): 792–4. PMID 8484118. doi:10.1126/science.8484118.
- ↑ Packer, L. (editor) (2002). Superoxide Dismutase: 349 (Methods in Enzymology). Academic Press. ISBN 0-12-182252-4.
- ↑ Heinrich, Peter; Löffler, Georg; Petrides, Petro E. (2006). Biochemie und Pathobiochemie (in German). Berlin: Springer. p. 123. ISBN 3-540-32680-4.
- ↑ Barondeau, D. P.; Kassmann, C. J.; Bruns, C. K.; Tainer, J. A.; Getzoff, E. D. (2004). "Nickel superoxide dismutase structure and mechanism". Biochemistry. 43 (25): 8038–8047. PMID 15209499. doi:10.1021/bi0496081.
- ↑ Parkin, Alison (2014). "Chapter 5. Understanding and Harnessing Hydrogenases, Biological Dihydrogen Catalysts". In Kroneck, Peter M. H.; Sosa Torres, Martha E. The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment. Metal Ions in Life Sciences. 14. Springer. pp. 99–124. doi:10.1007/978-94-017-9269-1_5.
- ↑ Pyle, A. M. (1993-08-06). "Ribozymes: a distinct class of metalloenzymes". Science. 261 (5122): 709–714. ISSN 0036-8075. PMID 7688142. doi:10.1126/science.7688142.
- ↑ Shan, Shu-ou; Yoshida, Aiichiro; Sun, Sengen; Piccirilli, Joseph A.; Herschlag, Daniel (1999-10-26). "Three metal ions at the active site of the Tetrahymena group I ribozyme". Proceedings of the National Academy of Sciences of the United States of America. 96 (22): 12299–12304. ISSN 0027-8424. PMC 22911 . PMID 10535916. doi:10.1073/pnas.96.22.12299.
- ↑ Weinberg, Zasha; Kim, Peter B.; Chen, Tony H.; Li, Sanshu; Harris, Kimberly A.; Lünse, Christina E.; Breaker, Ronald R. (2015-08-01). "New classes of self-cleaving ribozymes revealed by comparative genomics analysis". Nature Chemical Biology. 11 (8): 606–610. ISSN 1552-4450. PMC 4509812 . PMID 26167874. doi:10.1038/nchembio.1846.
- ↑ Breaker, R. R.; Joyce, G. F. (1994-12-01). "A DNA enzyme that cleaves RNA". Chemistry & Biology. 1 (4): 223–229. ISSN 1074-5521. PMID 9383394. doi:10.1016/1074-5521(94)90014-0.
- ↑ Silverman, Scott K. (2015-05-19). "Pursuing DNA catalysts for protein modification". Accounts of Chemical Research. 48 (5): 1369–1379. ISSN 1520-4898. PMC 4439366 . PMID 25939889. doi:10.1021/acs.accounts.5b00090.
- ↑ Santoro, Stephen W.; Joyce, Gerald F. (1997-04-29). "A general purpose RNA-cleaving DNA enzyme". Proceedings of the National Academy of Sciences. 94 (9): 4262–4266. ISSN 0027-8424. PMC 20710 . PMID 9113977. doi:10.1073/pnas.94.9.4262.
- ↑ Breaker, Ronald R.; Joyce, Gerald F. (1994-01-12). "A DNA enzyme that cleaves RNA". Chemistry & Biology. 1 (4): 223–229. ISSN 1074-5521. PMID 9383394. doi:10.1016/1074-5521(94)90014-0.
- ↑ Liu, Juewen; Brown, Andrea K.; Meng, Xiangli; Cropek, Donald M.; Istok, Jonathan D.; Watson, David B.; Lu, Yi (2007-02-13). "A catalytic beacon sensor for uranium with parts-per-trillion sensitivity and millionfold selectivity". Proceedings of the National Academy of Sciences. 104 (7): 2056–2061. ISSN 0027-8424. PMC 1892917 . PMID 17284609. doi:10.1073/pnas.0607875104.
- ↑ Torabi, Seyed-Fakhreddin; Wu, Peiwen; McGhee, Claire E.; Chen, Lu; Hwang, Kevin; Zheng, Nan; Cheng, Jianjun; Lu, Yi (2015-05-12). "In vitro selection of a sodium-specific DNAzyme and its application in intracellular sensing". Proceedings of the National Academy of Sciences. 112 (19): 5903–5908. ISSN 0027-8424. PMC 4434688 . PMID 25918425. doi:10.1073/pnas.1420361112.
- ↑ Stevens, F. C. (1983). "Calmodulin: an introduction". Can. J. Biochem. Cell Biol. 61 (8): 906–10. PMID 6313166. doi:10.1139/o83-115.
- ↑ Chin, D.; Means, A. R. (2000). "Calmodulin: a prototypical calcium sensor". Trends Cell Biol. 10 (8): 322–8. PMID 10884684. doi:10.1016/S0962-8924(00)01800-6.
- ↑ Berg, J. M. (1990). "Zinc finger domains: hypotheses and current knowledge". Annu Rev Biophys Biophys Chem. 19 (1): 405–421. PMID 2114117. doi:10.1146/annurev.bb.19.060190.002201.
- ↑ Jeoung, Jae-Hun; Fesseler, Jochen; Goetzl, Sebastian; Dobbek, Holger (2014). "Chapter 3. Carbon Monoxide. Toxic Gas and Fuel for Anaerobes and Aerobes: Carbon Monoxide Dehydrogenases". In Kroneck, Peter M. H.; Sosa Torres, Martha E. The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment. Metal Ions in Life Sciences. 14. Springer. pp. 37–69. doi:10.1007/978-94-017-9269-1_3.
- ↑ Aoki, Katsuyuki; Murayama, Kazutaka; Hu, Ning-Hai (2017). "Chapter 7. Solid State Structures of Lead Complexes with Relevance for Biological Systems". In Astrid, S.; Helmut, S.; Sigel, R. K. O. Lead: Its Effects on Environment and Health. Metal Ions in Life Sciences. 17. de Gruyter. pp. 123–200. doi:10.1515/9783110434330-007.
- ↑ Romani, Andrea M. P. (2013). "Chapter 4. Magnesium Homeostasis in Mammalian Cells". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-1_4.
- ↑ Roth, Jerome; Ponzoni, Silvia; Aschner, Michael (2013). "Chapter 6. Manganese Homeostasis and Transport". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-1_6.
- ↑ Dlouhy, Adrienne C.; Outten, Caryn E. (2013). "Chapter 8. The Iron Metallome in Eukaryotic Organisms". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-1_8.
- ↑ Cracan, Valentin; Banerjee, Ruma (2013). "Chapter 10 Cobalt and Corrinoid Transport and Biochemistry". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-10_10.
- ↑ Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O., eds. (2008). Nickel and Its Surprising Impact in Nature. Metal Ions in Life Sciences. 2. Wiley. ISBN 978-0-470-01671-8.
- ↑ Sydor, Andrew M.; Zambie, Deborah B. (2013). "Chapter 11. Nickel Metallomics: General Themes Guiding Nickel Homeostasis". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-10_11.
- ↑ Vest, Katherine E.; Hashemi, Hayaa F.; Cobine, Paul A. (2013). "Chapter 13. The Copper Metallome in Eukaryotic Cells". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-10_12.
- ↑ Maret, Wolfgang (2013). "Chapter 14 Zinc and the Zinc Proteome". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-10_14.
- ↑ Peackock, Anna F.A.; Pecoraro, Vincent (2013). "Chapter 10. Natural and artificial proteins containing cadmium". In Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O. Cadmium: From Toxicology to Essentiality. Metal Ions in Life Sciences. 11. Springer. pp. 303–337. doi:10.1007/978-94-007-5179-8_10.
- ↑ Freisinger, Elsa F.A.; Vasac, Milan (2013). "Chapter 11. Cadmium in Metallothioneins". In Sigel, Astrid; Sigel, Helmut; Sigel, Roland K. O. Cadmium: From Toxicology to Essentiality. Metal Ions in Life Sciences. 11. Springer. pp. 339–372. doi:10.1007/978-94-007-5179-8_11.
- ↑ Mendel, Ralf R. (2013). "Chapter 15. Metabolism of Molybdenum". In Banci, Lucia. Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. ISBN 978-94-007-5561-1. ISSN 1868-0402. doi:10.1007/978-94-007-5561-10_15.
- ↑ ten Brink, Felix (2014). "Chapter 2. Living on acetylene. A Primordial Energy Source". In Kroneck, Peter M. H.; Sosa Torres, Martha E. The Metal-Driven Biogeochemistry of Gaseous Compounds in the Environment. Metal Ions in Life Sciences. 14. Springer. pp. 15–35. doi:10.1007/978-94-017-9269-1_2.
External links
| Wikimedia Commons has media related to Metalloproteins. |
- Metalloprotein at the US National Library of Medicine Medical Subject Headings (MeSH)
- CATHERINE DRENNAN'S SEMINAR: SNAPSHOTS OF METALLOPROTEINS