Metalation
Metalation (Alt. Spelling: Metallation) is a chemical reaction which involves the bonding of a metal atom to what is typically an organic molecule to form a new compound. This reaction usually involves the replacement of a hydrogen atom in an organic molecule with a metal atom to form an organometallic compound. In the laboratory, metalation is commonly used to activate organic molecules during the formation of C—X bonds (where X is typically Carbon, Oxygen, or Nitrogen), which are necessary for the synthesis of many organic molecules.
In synthesis, metallated reagents are typically involved in nucleophilic substitution, single-electron-transfer (SET), and redox chemistry with functional groups on other molecules (including but not limited to ketones, aldehydes, and alkyl halides). Metallated molecules may also participate in acid-base chemistry, with one organometallic reagent deprotonating an organic molecule to create a new organometallic reagent.
The most common classes of metallated compounds are organolithium reagents and Gringard reagents. However, other organometallic compounds — such as organozinc compounds — also experience common use in both laboratory and industrial applications.
History
Metalation was first observed in the laboratory by Edward Frankland during a synthesis of diethylzinc in 1849.[1] While this development eventually led to the development of organometallic compounds of other metals,[2] these compounds saw little use in the laboratory because of their expense and (in the case of organozinc compounds) their highly pyrophoric nature. Metalation reactions (particularly in the form of transmetalation) only began to see more widespread use in synthetic laboratories after François Auguste Victor Grignard’s synthesized organomagnesium halides directly from metallic magnesium and organic halides.[3] These newfound organomagnesium reagents' extreme versatility in organic sythesis caused metalation to see widespread use in laboratory science.[4] Organolithium reagents were synthesized for the first time in 1917 by Schlenk and Holtz,[5] though these reagents did not see widespread use as metallating agents or reagents in organic synthesis until Karl Ziegler, Henry Gilman, and Georg Wittig — among others — developed synthetic methods that improved upon this initial synthesis.[6] After these improvements in synthesis came to be known, interest in the compounds increased significantly, as they are generally more reactive than organomagnesium compounds. The first use of an organolithium reagent as a metalation reagent occurred in 1928, with Schlenk and Bergmann's metalation of fluorene with ethyllithium.[7]
Reactivity and Applications
Most simple metallated compounds are commercially available in both the solid and solution phases, with solution phase metallated compounds available in a wide range of solvents and concentrations. These compounds may also be created in the laboratory as an in situ synthetic intermediate or separately in solution.
Reactivity of Metallated Compounds
The large difference in electronegativity between the carbon atom and metal atoms in most metallated compounds causes the resultant Carbon—Metal bond to be highly polar. The high polarity of the bond — and the resultantly high electron density around the metallated carbon atom — causes the bond's electronic character to strongly resemble that of an ionic bond. This makes metallated reagents generally good nucleophiles, and strong bases.
Metallated compounds are most commonly used in organic synthesis, where they act as nucleophiles in nucleophilic substitution reactions, strong bases in deprotonation reactions, initiators in polymerization reactions, and starting materials for the creation of other metallated compounds in transmetalation reactions.
Sterically hindered metallated compounds, such as n-Butyllithium complexes, are often used as superbases or polymerization initiators because their steric bulk hinders the compound's ability to approach nucleophiles at a distance short enough for nucleophilic attack. Metallated compounds without a high degree of steric bulk, such as methyllithium or alkyl magnesium halides, are more commonly used as nucleophiles or transmetallation reagents — though these compounds' high basicity often requires the protection of basic functional groups found on organic molecules.
Mechanism
Metalation is commonly used to synthesize complex organometallic reagents, such as alkynylithium reagents, from complex hydrocarbon molecules that possess acidic hydrogens. For both intermolecular and intramolecular metalation, the reaction occurs via the acid-base functionalization of the C-H bond by a Metal (M) — Base (B) pair according to the general scheme below.

The relative stability of the final products of this reaction determines whether or not this reaction is reversible, while the relative acidity of the C-H bonds present in the metallated molecule will determine the metalation location of the newly formed organometallic reagent.
Metalation was first proposed to follow a Concerted Metalation-Deprotonation (CMD) mechanism by Winstein and Traylor in 1955, postulating such on the basis of mercury’s electrophilicity during the acetolysis of diphenylmercury in acetic acid.[8] Later mechanistic studies support the existence of this mechanism for both intermolecular and intramolecular metalation reactions generating organometallic compounds. The commonly accepted mechanism is depicted below, with the metalation of a primary hydrogen given as an example.[9]

Transmetalation
(See: Transmetalation)
Transmetalation involves the exchange of two metals between organic molecules by a redox exchange mechanism. For example, transmetalations often form a reaction between an organolithium reagent and a metal salt.
Organolithium Reagent
(See: Organolithium reagent)
When synthesizing simple organolithium reagents, the reduction of one equivalent of a simple alkyl or aryl halide with two equivalents of Lithium metal produces one equivalent of a simple alkyl- or aryl-lithium and one equivalent of lithium halide with good yield.[10]

This reaction is known to proceed via a radical pathway that is likely initiated through a single-electron-transfer mechanism of the type shown below.[11]
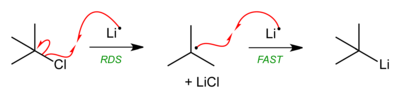
Magnesium similarly metalates organohalides to give Grignard reagents.
References
- ↑ Frankland, E. (1849). "Ueber die Isolirung der organischen Radicale". European Journal of Organic Chemistry. 71 (2): 171–213.
- ↑ Johnson, W.C. (1939). "Die Chemie der Metall-Organischen Verbindungen (Krause, Erich; Grosse, A. V.)". J. Chem. Education. 16(3): 148.
- ↑ Grignard, V. (1900). "Sur quelques nouvelles combinaisons organométaliques du magnésium et leur application à des synthèses d'alcools et d'hydrocabures". Compt. Rend. 130: 1322–25.
- ↑ Eisch, John J. (2002). "Henry Gilman: American Pioneer in the Rise of Organometallic Chemistry in Modern Science and Technology". Organometallics. 21 (25): 5439–5463.
- ↑ Schlenk, W.; Holtz, J. (1917). "Über die einfachsten metallorganischen Alkaliverbindungen". European Journal of Inorganic Chemistry. 50 (1): 262–274.
- ↑ Gilman, H.; Zoellner, E. A.; Selby, W. M. (1932). "AN IMPROVED PROCEDURE FOR THE PREPARATION OF ORGANOLITHIUM COMPOUNDS". J. Am. Chem. 54 (5): 1957–1962.
- ↑ Schlenk and Bergmann, Ann., 463, 192, 1928
- ↑ Winstein, S.; Traylor, T.G. (1955). "Mechanisms of Reaction of Organomercurials. II. Electrophilic Substitution on Saturated Carbon. Acetolysis of Dialkylmercury Compounds.". J. Am. Chem. 77 (14): 3747–3752.
- ↑ Lapointe, D.; Fagnou, K. (2010). "Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway". The Chemical Society of Japan Chemistry Letters. 39 (11): 1118–1126.
- ↑ "Organometallics in Organic Synthesis", Schlosser, M., Ed, Wiley: New York, 1994. ISBN 0-471-93637-5
- ↑ Bailey, William F.; Patricia, Jeffrey J. (1988). "The mechanism of the lithium - halogen Interchange reaction : a review of the literature". Journal of Organometallic Chemistry. 352 (1-2): 1–46.