MALBAC
Multiple Annealing and Looping Based Amplification Cycles (a.k.a. MALBAC) is a quasilinear whole genome amplification method. Unlike conventional DNA amplification methods that are non-linear or exponential (in each cycle, DNA copied can serve as template for subsequent cycles), MALBAC utilizes special primers that allow amplicons to have complementary ends and therefore to loop, preventing DNA from being copied exponentially. This results in amplification of only the original genomic DNA and therefore reduces amplification bias. MALBAC is “used to create overlapped shotgun amplicons covering most of the genome”.[1] For next generation sequencing, MALBAC is followed by regular PCR which is used to further amplify amplicons.
Technological platform
Prior to MALBAC, a single cell is isolated by various methods including laser capture microdissection, microfluidic devices, flow cytometry, or micro pipetting, then lysed. MALBAC single-cell whole-genome amplification involves 5 cycles of quenching, extending, melting, and looping.
MALBAC primers
The major advantage of MALBAC is that DNA is amplified almost linearly. The utilization of specialized primers enables looping of amplicons which then prevents them from being further amplified in subsequent cycles of MALBAC. These primers are 35 nucleotides long, with 8 variable nucleotides that hybridize to the templates and 27 common nucleotides.[1] The common nucleotide sequence is GTG AGT GAT GGT TGA GGT AGT GTG GAG. The 8 variable nucleotides anneal randomly to the single stranded genomic DNA molecule. After one extension, semi-amplicon, an amplicon containing the common nucleotide sequence on only the 5’ end, is made. This semi-amplicon is used as a template for another round of extension, which then results in a full-amplicon, an amplicon where the 3’ end is complementary to the sequence on the 5’ end.
Strand displacement
MALBAC primers have variable components which allow them to randomly bind to the template DNA. This means that on a single fragment at any cycle, there could be multiple primers annealed to the fragment. A DNA polymerase such as one derived from Bacillus stearothermophilus (Bst polymerase) is able to displace the 5’ end of another upstream strand growing in the same direction.[2]
Error rate
Bst DNA polymerase has an error rate of 1/10000 bases.[3]
Experimental workflow
_Workflow.jpg)
- Single-cell isolation and lysis – pg of genomic DNA fragments (10 to 100 kb) isolated from a single-cell are used as templates.
- Melting – At 94 °C, double-stranded DNA molecules are melted into single stranded forms.
- Quenching – After melting, the reaction is immediately quenched to 0 °C, and MALBAC primers are added to the reaction.
- Extension – Bst DNA Polymerase (Large Fragment) extends the primers at 65 °C for 2 mins, creating semi-amplicons.
- Melting – The reaction is heated to back to 94 °C to separate the semi-amplicon from the genomic DNA template.
- Quenching - The reaction is quickly quenched at 0 °C, and followed by the addition of the same polymerase mix. The MALBAC primers efficiently bind to both semi-amplicons and genomic DNA template.
- Extension - Bst DNA Polymerase (Large Fragment) extends the primers at 65 °C for 2 mins. At this step, full-amplicons are made for those that used semi-amplicons as templates, and also semi-amplicons are made for those that used the genomic DNA template as templates.
- Melting – The reaction is heated to 94 °C to separate the amplicons from the template.
- Looping – For full amplicons, the 3’ end sequence is now complementary to the 5’ end. At 58 °C, the two ends hybridize forming a looped DNA. This prevents the full amplicon from being used as a template in subsequent MALBAC cycles.
- Repeat steps 6-9 five times – 5 cycles of linear MALBAC amplification.
- Regular PCR – The MALBAC product is further amplified by PCR. By using the 27 common nucleotides as primers, only the full amplicons are amplified.
At the end of PCR, picograms of genetic material is amplified to microgram of DNA, yielding enough DNA to be sequenced.
Applications
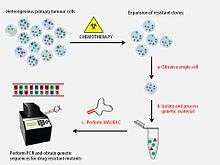
MALBAC offers an unbiased approach to the amplification of DNA from a single cell. This method of single cell sequencing has a vast number of applications, many of which have yet to be exploited. MALBAC may aid in the analysis of forensic specimens, in pre-natal screening for genetic diseases, in understanding the development of reproductive cells, or in elucidating the complexity of a tumour.[1][4] At its foundation, this technology allows researchers to observe the frequency with which mutations accumulate in single cells.[1] Moreover, it permits the detection of chromosomal abnormalities and gene copy number variations (CNVs) within and between cells, and further facilitates the detection of uncommon mutations that result in single nucleotide polymorphisms (SNPs).[1]
In the field of cancer research, MALBAC has many applications. It may be used to examine intratumor heterogeneity, to identify genes which may confer an aggressive or metastatic phenotype, or to evaluate the potential for a tumour to develop drug resistance.[4][5] A pioneering application of MALBAC was published in a December 2012 issue of Science and described the use of this technology to measure the mutation rate of the colon cancer cell line SW4802.[1] By sequencing the amplified DNA of three kindred colon cancer cells in parallel with unrelated colon cancer cells from a different lineage, SNPs were identified with no false positives detected.[1] It was also observed that purine-pyrimidine transversions occurred at a high frequency among the SNPs.[1] The characterization of copy number and single nucleotide variations of single colon cancer cells highlighted the heterogeneity present within a tumour.[1]
MALBAC has been applied as a method to examine the genetic diversity amongst reproductive cells. By sequencing the genomes of 99 individual human sperm cells from an anonymous donor, MALBAC was used to examine genetic recombination events involving single gametes and ultimately provide insight into the dynamics of genetic recombination and its contribution to male infertility.[6] Additionally, within an individual sperm, MALBAC identified duplicated or missing chromosomes, as well as SNPs or CNVs which could negatively affect fertility.[6]
Advantages
MALBAC has resulted in many significant advances over other single cell sequencing techniques, foremost that it can report 93% of the genome of a single human cell.[1] Some advantages of this technology include reduced amplification bias and increased genome coverage, the requirement for very little template DNA, and low rates of false positive and false negative mutations.[4][6]
Reduces amplification bias and increases genome coverage
MALBAC is a form of whole genome sequencing which reduces the bias associated with exponential PCR amplification by using a quasilinear phase of pre-amplification.[1] MALBAC utilizes five cycles of pre-amplification and primers containing a 27 nucleotide common sequence and an 8 nucleotide variable sequence to produce fragments of amplified DNA (amplicons) which loop back on themselves to prevent additional copying and cross-hybridization.[1][7] These loops cannot be used as a template for amplification during MALBAC and therefore reduce the amplification bias commonly associated with the uneven exponential amplification of DNA fragments by polymerase chain reaction.[1] MALBAC has been described to have better amplification uniformity than other methods of single sequencing, such as multiple displacement amplification (MDA).[1][5] MDA does not utilize DNA looping and amplifies DNA in an exponential fashion, resulting in bias.[1] Accordingly, the amplification bias associated with other single cell sequencing methods results in low coverage of the genome.[1][5] The reduced bias associated with MALBAC has generated better genome sequence coverage than other single cell sequencing methods.
Requires very little template DNA
MALBAC can be used to amplify and subsequently sequence DNA when only one or a few cells are available, such as in the analysis of circulating tumour cells, pre-natal screens or forensic samples.[4][7] Only a small amount of starting template (picograms of DNA) is required to initiate the process, and therefore it is an ideal method for the sequencing of a single human cell.[1]
Low incidence of false positive and false negative mutations
Single cell sequencing often has a high rate of false negative mutations.[1] A false negative mutation rate is defined as the probability of not detecting a real mutation, and this may occur due to amplification bias resulting from the loss, or drop-out, of an allele.[8] The sequence coverage uniformity of MALBAC in comparison to other single cell sequencing techniques has enhanced the detection of SNPs and reduced allele dropout rate.[1] Allelic dropout rate increases when an allele of a heterozygote fails to amplify resulting in identification of a ‘false homozygote.’ This may occur due to low concentration of DNA template, or the uneven amplification of template resulting in one allele of a heterozygote being copied more than the other.[8] The allele dropout rate of MALBAC has been shown to be much lower (approximately 1%) compared to MDA which is approximately 65%. In contrast to MDA which has been shown to have a 41% SNP detection efficiency in comparison with bulk sequencing, MALBAC has been reported to have SNP detection deficiency of 76%.[1] MALBAC has also been reported to have a low false positive rate. False positive mutations generated by MALBAC largely result from errors introduced by DNA polymerase during the first cycle of amplification that are further propagated during subsequent cycles. This false positive rate can be eliminated by sequencing 2-3 cells within a lineage derived from a single cell to verify the presence of a SNP, and by eliminating sequencing and amplification errors by sequencing unrelated cells from a separate lineage.[1]
Limitations
- Due to the requirement for a low quantity of template DNA, contamination of target DNA by the operator or environment can potentially confound sequencing results.[1]
- In order to completely rule out false positives, it is necessary to compare the cell sequencing results to those obtained from 2-3 cells within the same lineage, as well as to cells from an unrelated lineage.[1]
- DNA polymerase used to amplify the template DNA is error prone and can introduce sequencing errors in the first cycle of MALBAC which are subsequently propagated.[1][4]
- Genome coverage at a single cell level using MALBAC is less uniform than bulk sequencing. Although MALBAC has improved the detection efficiency of single cell sequencing, it is unable to detect approximately one third of SNPs compared to bulk sequencing.[1][4]
External links
- NCBI database
- Yikon Genomics:Whole Genome Amplification (WGA) and Multiple Annealing and Looping-based Amplification Cycles (MALBAC)
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 Zong, C.; Lu, S.; Chapman, A.R.; Xie, S. (2012). "Genome-wide detection of single-nucleotide and copy-number variations of a single human cell." Science 338, 1622. DOI: 10.1126/science.1229164. PMID 23258894
- ↑ "Aviel-Ronen, S.; Qi Zhu, C.; Coe, B.P.; Liu, N.; Watson, S.K.; Lam, W.L.; Tsao, M.S. (2006) "Large fragment Bst DNA polymerase for whole genome amplification of DNA from formalin-fixed paraffin-embedded tissues." BMC Genomics 7. DOI:10.1186/1471-2164-7-312. PMID 17156491
- ↑ CURRENT PROTOCOLS IN MOLECULAR BIOLOGY, Ausubel, F.M. et al. Vol. I., John Wiley & Songs, Inc. 1995. Pp 7.4.18.
- 1 2 3 4 5 6 Baker, M. (2012). "Method offers blueprint of a single human cell." Nature News. doi:10.1038/nature.2012.12088
- 1 2 3 Lu, S.; Zong, C.; Fan, W.; Yang, M.; Li, J.; Chapman, A.R.; Zhu, P.; Hu, X.; Xu, L.; Yan, L.; Bai, F.; Qiao, J.; Tang, F.; Li, R.; Xie, S. (2012). "Probing meiotic recombination and aneuploidy of single sperm cells by whole-genome sequencing." Science 338, 1627. DOI: 10.1126/science.1229112. PMID 23258895
- 1 2 Reuell, P. (2013). "One cell is all you need – innovative technique can sequence entire genome from single cell." Harvard Gazette. http://news.harvard.edu/gazette/story/2013/01/one-cell-is-all-you-need/
- 1 2 Miller, C.R.; Joyce, P.; Waits, L.P. (2002). "Assessing allelic dropout and genotype reliability using maximum likelihood." Genetics 160 (1) 357-366. PMID 11805071