Langer–Giedion syndrome
| Langer–Giedion syndrome | |
|---|---|
| Synonyms | Deletion 8q24.1,Monosomy 8q24.1 |
 | |
| A person showing the typical features of Langer-Giedion syndrome | |
| Classification and external resources | |
| Specialty | medical genetics |
| ICD-10 | ICD-10: Q87.8 |
| OMIM | 150230 |
| DiseasesDB | 31949 |
| MeSH | D015826 |
| Orphanet | 502 |
Langer–Giedion syndrome (LGS), also called trichorhinophalangeal syndrome type II (TRPS2) or LGCR (for "Langer-Giedion Chromosome Region"),[1][2] is a very uncommon autosomal dominant genetic disorder caused by a deletion of chromosomal material. It is named after the two doctors who undertook the main research into the condition in the 1960s. Diagnosis is usually made at birth or in early childhood.
Symptoms
The features associated with this condition include: mild to moderate learning difficulties, short stature, unique facial features, small head and skeletal abnormalities including bony growths projecting from the surfaces of bones.[3] Typically individuals with Langer–Giedion syndrome have fine scalp hair, ears that may be large or prominent, broad eyebrows, deep-set eyes, a bulbous nose, long narrow upper lip, and missing teeth.
Cause
Deletion 8q23.2 to q24.1.[2]
It involves a loss of TRPS1 and EXT1.
Genetics
The syndrome occurs when a small piece of chromosome 8's long arm, which contains a number of genes, is missing. The loss of these genes is responsible for some of the overall characteristics of Langer–Giedion syndrome.
The missing portion of the chromosome is 8q23-q24. This region includes the genes TRPS1 and EXT1.
Diagnosis
Diagnosis is based on clinical findings and can be confirmed by cytogenetic testing, when the deletion is in an average of 5 Mb (millions of base pairs). Nowadays is a common practice to run an aCHG (array chromosome hybridization genome) study on peripheral blood of the patient, in order to limit the extent of the loss of the genomic area, and the deleted genes.
Treatment
While no genetic syndrome is capable of being cured, treatments are available for some symptoms. External fixators have been used for limbic and facial reconstructions.
Image gallery
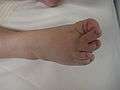 The right foot of a person with Langer–Giedion syndrome showing the characteristic features
The right foot of a person with Langer–Giedion syndrome showing the characteristic features Hands of a person with Langer–Giedion syndrome showing the characteristic short fingers.
Hands of a person with Langer–Giedion syndrome showing the characteristic short fingers.
See also
References
- ↑ Online Mendelian Inheritance in Man (OMIM) 150230
- 1 2 McBrien, J.; Crolla, J. A.; Huang, S.; Kelleher, J.; Gleeson, J.; Lynch, S. A. (June 2008). "Further case of microdeletion of 8q24 with phenotype overlapping Langer–Giedion withoutTRPS1 deletion". American Journal of Medical Genetics Part A. 146A (12): 1587–1592. PMID 18478595. doi:10.1002/ajmg.a.32347.
- ↑ Devidayal; Marwaha RK (February 2006). "Langer-Giedion Syndrome" (PDF). Indian Pediatrics. 43 (2): 174–175. PMID 16528117.
- ↑ http://www.omim.org/entry/150230
External links
- Trichorhinophalangeal syndrome type 2 at NIH's Office of Rare Diseases