Facioscapulohumeral muscular dystrophy
| Facioscapulohumeral muscular dystrophy | |
|---|---|
| Timelapse of DUX4 being expressed in FSHD Muscle Cells[1] | |
| Classification and external resources | |
| Specialty | neurology |
| ICD-10 | G71.0 |
| ICD-9-CM | 359.1 |
| OMIM | 158900 158901 |
| DiseasesDB | 7247 |
| MedlinePlus | 000707 |
| eMedicine | neuro/133 |
| Patient UK | Facioscapulohumeral muscular dystrophy |
| MeSH | D020391 |
| GeneReviews | |
| Orphanet | 269 |
Facioscapulohumeral muscular dystrophy (FSHMD, FSHD or FSH)—originally named Landouzy-Dejerine[2]—is a usually autosomal dominant inherited form of muscular dystrophy (MD)[3] that initially affects the skeletal muscles of the face (facio), scapula (scapulo) and upper arms (humeral). FSHD is the third most common genetic disease of skeletal muscle. Orpha.net lists the prevalence as 4/100,000[4] while a 2014 population-based study in the Netherlands reported a significantly higher prevalence of 12 in 100,000.[5]
Symptoms may develop in early childhood and are usually noticeable in the teenage years, with 95% of affected individuals manifesting disease by age 20 years. A progressive skeletal muscle weakness usually develops in other areas of the body as well; often the weakness is asymmetrical. Life expectancy can be threatened by respiratory insufficiency, and up to 20% of affected individuals become severely disabled, requiring use of a wheel chair or mobility scooter. In a Dutch study, approximately 1% of patients required (nocturnal or diurnal) ventilatory support.[6] Non-muscular symptoms frequently associated with FSHD include subclinical sensorineural hearing loss and retinal telangiectasia. In more than 95% of known cases, the disease is associated with contraction of the D4Z4 repeat in the 4q35 subtelomeric region of Chromosome 4. Seminal research published in August 2010 now shows the disease requires a second mechanism, which for the first time provides a unifying theory for its underlying genetics. The second mechanism is a "toxic gain of function" of the DUX4 gene, which is the first time in genetic research that a "dead gene" has been found to "wake up" and cause disease.[7][8]
Building on the 2010 unified theory of FSHD, researchers in 2014 published the first proposed pathophysiology definition of the disease and four viable therapeutic targets for possible intervention points.[9]
Symptoms
Because of the extreme variability of the disease, an authoritative and scientifically confirmed set of symptoms does not yet exist. The prevalence is widely placed at 1/20,000, but the exact prevalence is not known. A November 2008 report from Orpha.net, an organization backed by the Institut National de la Santé et de la Recherche Médicale (INSERM), listed a prevalence of 7/100,000, but the May 2014 version of this report places the prevalence at 4/100,000.[4] A 2014 population-based study in the Netherlands reported a significantly higher prevalence of 12 in 100,000.[4]
Symptoms:
- Facial muscle weakness (eyelid drooping, inability to whistle, decreased facial expression, depressed or angry facial expression, difficulty pronouncing the letters M, B, and P)
- Shoulder weakness (difficulty working with the arms raised, sloping shoulder)
- Hearing loss
- Abnormal heart rhythm
- Unequal weakening of the biceps, triceps, deltoids, and lower arm muscles
- Loss of strength in abdominal muscles (causing a protuberant abdomen and lumbar lordosis) and eventual progression to the legs
- Foot drop
Genetics

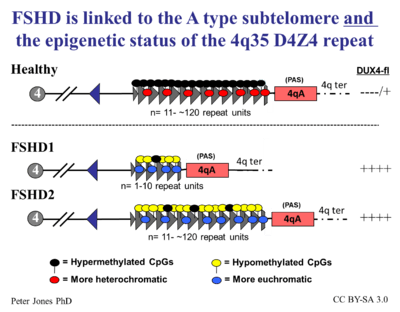
FSHD Type 1 (also called FSHMD1A) (4q35 deletion)
More than 95% of cases of FSHD are associated with the deletion of integral copies of a tandemly repeated 3.2kb unit (D4Z4 repeat) at the subtelomeric region 4q35 on Chromosome 4 of the human genome, of which a normal chromosome includes between 11-150 repetitions of D4Z4.[10] There are both heterochromatin and euchromatin structures within D4Z4 and one putative gene called DUX4.[10][11] Inheritance is autosomal dominant, though up to one-third of the cases appear to be from de novo (new) mutations. The heterochromatin is specifically lost in the deletions of FSHD while the euchromatin structures remain.[10] If the entire region is removed, there are birth defects, but no specific defects on skeletal muscle. Individuals appear to require the existence of 11 or fewer repeat units to be at risk for FSHD.
In addition, a few cases of FSHD are the result of rearrangements between subtelomeric chromosome 4q and a subtelomeric region of 10q. This location contains a tandem repeat structure highly homologous to 4q35.[12] Disease occurs when the translocation results in a critical loss of tandem repeats to the 4q site.
FSHD Type 2
A large family was reported with a phenotype indistinguishable from FSHD in which no pathological changes at the 4q site or translocation of 4q-10q are found.[13][14] It had been suggested that this may be due to limitations in the available tests.[15]
In 2012, a majority of FSHD2 cases were reported linked to mutations in the SMCHD1 gene on chromosome 18. This leads to substantially reduced levels of SMCHD1 protein, and subsequently, hypomethylation of the 4q D4Z4 region. The FSHD2 phenotype arises in individuals who inherited both the SMCHD1 mutations plus a normal sized D4Z4 region on a permissive 4qA allele. This establishes a genetic/mechanistic intersection of FSHD1 and FSHD2.[16]
A Unifying Theory
On 19 August 2010, a paper entitled A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy was published in Science showing that the candidate gene DUX4 undergoes a "toxic gain of function" as a result of single nucleotide polymorphisms in the region distal to the last D4Z4 repeat. According to the research, this leads to a "canonical polyadenylation signal for transcripts derived from DUX4".[8] This is the first time in the history of genetics in which "junk" DNA has been shown to reanimate and cause disease. The documentation of the conditions under which the DUX4 gene became reanimated answered the question of why no one whose dead gene was repeated more than 10 times ever got FSHD but only some people with fewer than 10 copies did get the disease[7] Several organizations including the New York Times highlighted this research (See MDA, FSH Society, University of Rochester, NYT).
Dr. Francis Collins, who oversaw the first sequencing of the Human Genome with the National Institutes of Health stated:[7]
“If we were thinking of a collection of the genome’s greatest hits, this would go on the list,”
Daniel Perez, co-founder, President and CEO of the FSH Society hailed the new findings saying:
"This is a long-sought explanation of the exact biological workings of a disease that affects an estimated one in 14,000 or 22,100 Americans and 490,000 worldwide,” he said, adding that this discovery “creates an enormous opportunity for research to develop ways to prevent or treat FSHD.”
The MDA stated that:
"The new findings will make it easier to diagnose FSHD in someone with symptoms and predict who will develop the disease in someone without symptoms. Now, the hunt is on for which proteins or genetic instructions (RNA) cause the problem for muscle tissue in FSHD."
Quoted in the University of Rochester press release, one of the report's co-authors, Silvère van der Maarel of the University of Leiden, stated that
“It is amazing to realize that a long and frustrating journey of almost two decades now culminates in the identification of a single small DNA variant that differs between patients and people without the disease. We finally have a target that we can go after.”
The original identification of the D4Z4 deletions was found in 1992. This research now shows that a second mechanism is needed for FSHD to be present and that the remaining versions of the DUX4 become more active (open for transcription) because the DNA at the tip of chromosome 4 is less tightly coiled as a result of the deletions.
Chronology of Important FSHD-related Genetic Research
- Landouzy and Dejerine describe a form of childhood progressive muscle atrophy with a characteristic involvement of facial muscles and distinct from pseudohypertrophic (Duchenne’s MD) and spinal muscle atrophy in adults.[17]
1886
- Landouzy and Dejerine describe progressive muscular atrophy of the scapulo-humeral type.[18]
1950
- Tyler and Stephens study 1249 individuals from a single kindred with FSHD traced to a single ancestor and describe a typical Mendelian inheritance pattern with complete penetrance and highly variable expression. The term facioscapulohumeral dystrophy is introduced.[19]
1982
- Padberg provides the first linkage studies to determine the genetic locus for FSHD in his seminal thesis "Facioscapulohumeral disease."[20]
1987
- The complete sequence of the Dystrophin gene (Duchenne’s MD) is determined.[21]
1991
- The genetic defect in FSHD is linked to a region (4q35) near the tip of the long arm of chromosome 4.[22]
1992
- FSHD, in both familial and de novo cases, is found to be linked to a recombination event that reduces the size of 4q EcoR1 fragment to < 28 kb (50–300 kb normally).[23]
1993
- 4q EcoR1 fragments are found to contain tandem arrangement of multiple 3.3-kb units (D4Z4), and FSHD is associated with the presence of < 11 D4Z4 units.[24]
- A study of seven families with FSHD reveals evidence of genetic heterogeneity in FSHD.[25]
1994
- The heterochromatic structure of 4q35 is recognized as a factor that may affect the expression of FSHD, possibly via position-effect variegation.[26]
- DNA sequencing within D4Z4 units shows they contain an open reading frame corresponding to two homeobox domains, but investigators conclude that D4Z4 is unlikely to code for a functional transcript.[26][27]
1995
- The terms FSHD1A and FSHD1B are introduced to describe 4q-linked and non-4q-linked forms of the disease.[28]
1996
1998
- Monozygotic twins are identified with identical 23 kb EcoR1 fragments but vastly different clinical expression of FSHD.[30]
1999
- Complete sequencing of 4q35 D4Z4 units reveals a promoter region located 149 bp 5' from the open reading frame for the two homeobox domains, indicating a gene that encodes a protein of 391 amino acid protein (later corrected to 424 aa[31]), given the name DUX4.[32]
2001
- Investigators assessed the methylation state (heterochromatin is more highly methylated than euchromatin) of DNA in 4q35 D4Z4. An examination of SmaI, MluI, SacII, and EagI restriction fragments from multiple cell types, including skeletal muscle, revealed no evidence for hypomethylation in cells from FSHD1 patients relative to D4Z4 from unaffected control cells or relative to homologous D4Z4 sites on chromosome 10. However, in all instances, D4Z4 from sperm was hypomethylated relative to D4Z4 from somatic tissues.[33]
2002
- A polymorphic segment of 10 kb directly distal to D4Z4 is found to exist in two allelic forms, designated 4qA and 4qB. FSHD1 is associated solely with the 4qA allele.[34]
- Three genes (FRG1, FRG2, ANT1) located in the region just centromeric to D4Z4 on chromosome 4 are found in isolated muscle cells from individuals with FSHD at levels 10 to 60 times greater than normal, showing a linkage between D4Z4 contractions and altered expression of 4q35 genes.[35]
2003
- A further examination of DNA methylation in different 4q35 D4Z4 restriction fragments (BsaAI and FseI) showed significant hypomethylation at both sites for individuals with FSHD1, non-FSHD-expressing gene carriers, and individuals with phenotypic FSHD relative to unaffected controls.[36]
2004
- Contraction of the D4Z4 region on the 4qB allele to < 38 kb does not cause FSHD.[37]
2006
- Transgenic mice overexpressing FRG1 are shown to develop severe myopathy.[38]
2007
- The DUX4 open reading frame is found to have been conserved in the genome of primates for over 100 million years, supporting the likelihood that it encodes a required protein.[39]
- Researchers identify DUX4 mRNA in primary FSHD myoblasts and identify in D4Z4-transfected cells a DUX4 protein, the overexpression of which induces cell death.[31]
- DUX4 mRNA and protein expression are reported to increase in myoblasts from FSHD patients, compared to unaffected controls. Stable DUX4 mRNA is transcribed only from the most distal D4Z4 unit, which uses an intron and a polyadenylation signal provided by the flanking pLAM region. DUX4 protein is identified as a transcription factor, and evidence suggests overexpression of DUX4 is linked to an increase in the target paired-like homeodomain transcription factor 1 (PITX1).[40]
2009
- The terms FSHD1 and FSHD2 are introduced to describe D4Z4-deletion-linked and non-D4Z4-deletion-linked genetic forms, respectively. In FSHD1, hypomethylation is restricted to the short 4q allele, whereas FSHD2 is characterized by hypomethylation of both 4q and both 10q alleles.[41]
- Splicing and cleavage of the terminal (most telomeric) 4q D4Z4 DUX4 transcript in primary myoblasts and fibroblasts from FSHD patients is found to result in the generation of multiple RNAs, including small noncoding RNAs, antisense RNAs and capped mRNAs as new candidates for the pathophysiology of FSHD.[42]
2010
- A unifying genetic model of FSHD describes the requirement for FSHD-type deletions of D4Z4 to occur on a permissive allele containing a poly-A adenylation signal (PAS) in the pLAM1 region adjacent to the final D4Z4 unit. The non-permissive 4qB allele lacks a PAS, does not generate a stable DUX4 transcript, and is not linked to FSHD. The corresponding D4Z4 region on chromosome 10 (10q26) lacks a PAS altogether, and deletions in this region are not involved in FSHD.[43]
- DUX4 is found actively transcribed in skeletal muscle biopsies and primary myoblasts. FSHD-affected cells produce a full length transcript, DUX4-fl, whereas alternative splicing in unaffected individuals results in the production of a shorter, 3'-truncated transcript (DUX4-s). The low overall expression of both transcripts in muscle is attributed to relatively high expression in a small number of nuclei (~ 1 in 1000). Higher levels of DUX4 expression in human testis (~100 fold higher than skeletal muscle) suggest a developmental role for DUX4 in human development. Higher levels of DUX4-s (vs DUX4-fl) are shown to correlate with a greater degree of DUX-4 H3K9me3-methylation.[44]
2012
- Some, but not all, instances of FSHD2 are linked to mutations in the SMCHD1 gene on chromosome 18. This leads to substantially reduced levels of SMCHD1 protein, and subsequently, hypomethylation of the 4q D4Z4 region. The FSHD2 phenotype arises in individuals who inherited both the SMCHD1 mutations plus a normal sized D4Z4 region on a permissive 4qA allele. This establishes a genetic/mechanistic intersection of FSHD1 and FSHD2.[16]
- The prevalence of FSHD-like D4Z4 deletions on permissive alleles is significantly higher than the prevalence of FSHD in the general population, challenging the criteria for molecular diagnosis of FSHD.[45]
- When expressed in primary myoblasts, DUX4-fl acted as a transcriptional activator, producing a > 3-fold change in the expression of 710 genes.[46] A subsequent study using a larger number of samples identified DUX4-fl expression in myogenic cells and muscle tissue from unaffected relatives of FSHD patients, per se, is not sufficient to cause pathology, and that additional modifiers are determinants of disease progression.[47]
- A mechanism is proposed in which DBE-T (D4Z4 Regulatory Element transcript), a long non-coding RNA transcribed from a 4q35 region proximal to D4Z4, is expressed in FSHD, leading to the recruitment of the Trithorax group protein Ash1L, an increase in H3k36me2-methylation, and ultimately de-repression of 4q35 genes.[48]
2013
- Mutations in SMCHD1 are shown to act as a disease modifier, increasing the severity of FSHD1 in individuals who also exhibit a contraction of D4Z4.[49]
- Transgenic mice carrying D4Z4 arrays from an FSHD1 allele (with 2.5 D4Z4 units), although lacking an obvious FSHD-like skeletal muscle phenotype, are found to recapitulate important genetic expression patterns and epigenetic features of FSHD.[50]
2014
- DUX4-fl and downstream target genes are expressed in skeletal muscle biopsies and biopsy-derived cells of fetuses with FSHD-like D4Z4 arrays, indicating that molecular markers of FSHD are already expressed during fetal development.[51]
- In an open access article in Skeletal Muscle, researchers "review how the contributions from many labs over many years led to an understanding of a fundamentally new mechanism of human disease" and articulate how the unifying genetic model and subsequent research represent a "pivot-point in FSHD research, transitioning the field from discovery-oriented studies to translational studies aimed at developing therapies based on a sound model of disease pathophysiology." They describe the consensus mechanism of pathophysiology for FSHD as a "inefficient repeat-mediated epigenetic repression of the D4Z4 macrosatellite repeat array on chromosome 4, resulting in the variegated expression of the DUX4 retrogene, encoding a double-homeobox transcription factor, in skeletal muscle." [9]
FSH Society
In 1991 the FSH Society was founded by two individuals with FSHD, Daniel Perez and Stephen Jacobsen. The FSH Society raised funding to provide seed grants for FSHD research, advocated for the field to standardize the name of the disease as "facioscapulohumeral muscular dystrophy" and "FSHD", and co-wrote the MD-CARE Act, passed into law in 2001, which for the first time mandated federal resources, including National Institutes of Health funding, for all muscular dystrophies. The FSH Society has grown into the world's largest grassroots organization advocating for patient education and scientific and medical research.[52]
FSHD Foundation
In 2007 the FSHD Global Research Foundation was established to increase the amount of funding available to research bodies. The Foundation has identified 13 priority areas of interest for FSHD research.[53]
FSHD-EUROPE
In 2009 the FSHD-EUROPE was founded by European associations.[54]
Pathophysiology

In 2014, researchers undertook a "review [of] how the contributions from many labs over many years led to an understanding of a fundamentally new mechanism of human disease" and articulated how the unifying genetic model and subsequent research represent a "pivot-point in FSHD research, transitioning the field from discovery-oriented studies to translational studies aimed at developing therapies based on a sound model of disease pathophysiology." They proposed a consensus mechanism of pathophysiology for FSHD as a "inefficient repeat-mediated epigenetic repression of the D4Z4 macrosatellite repeat array on chromosome 4, resulting in the variegated expression of the DUX4 retrogene, encoding a double-homeobox transcription factor, in skeletal muscle." [9]
In more lay terms, the D4Z4 repeats (most people have about 200 or so) normally keep DUX4 repressed (the repeat-mediated repression). When there are drastically fewer repeats (approximately 10 or less) in addition to the small genetic change on Chromosome 4 called a haplotype polymorphism, DUX4 expresses itself (the inefficient repression component) via a complex set of mechanisms that make the genetic neighborhood around the DUX4 gene more conducive to gene expression (the epigenetic component). The figure on the right describes this process in detail.
Testing
Since the early 2000s, genetic testing that measures the size of the D4Z4 deletions on 4q35 has become the preferred mechanism for confirming the presence of FSHD. As of 2007, this test is considered highly accurate but is still performed by a limited set of labs in the US, such as Athena diagnostics under test code 405. However, because the test is expensive, patients and doctors may still rely on one or more of the following tests, all of which are far less accurate and specific than the genetic test:[55]
- Creatine kinase (CK) level: This test measures the Creatine kinase enzyme in the blood. Elevated levels of CK are related to muscle atrophy.
- electromyogram (EMG): This test measures the electrical activity in the muscle
- nerve conduction velocity (NCV): This test measures the how fast signals travel from one part of a nerve to another. The nerve signals are measured with surface electrodes (similar to those used for an electrocardiogram), and the test is only slightly uncomfortable.
- muscle biopsy: Through outpatient surgery a small piece of muscle is removed (usually from the arm or leg) and evaluated with a variety of biochemical tests. Researchers are attempting to match results of muscle biopsies with DNA tests to better understand how variations in the genome present themselves in tissue anomalies.
Therapies
- No approved therapies exist specifically for FSHD.
- Based on the consensus model of pathophysiology, researchers propose four approaches for therapeutic intervention:[9]
- enhance the epigenetic repression of the D4Z4
- target the DUX4 mRNA, including altering splicing or polyadenylation;
- block the activity of the DUX4 protein
- inhibit the DUX4-induced process, or processes, that leads to pathology.
- Physiotherapy could improve patients' functional status by providing therapeutic exercises.
- Occupational therapy can sometimes be used for ADL training and to help cope with new devices to make things easier.
- Several compounds aimed at increasing muscle mass have been pursued including:
- ACVR2B is a compound identified in 2005/2006 by researchers at Johns Hopkins. It increased muscle mass in a non-muscular dystrophy mouse by up to 60% in two weeks.
- MYO-029 (Stamulumab) is an experimental myostatin-inhibiting drug developed by Wyeth Pharmaceuticals for the treatment of muscular dystrophy. Myostatin is a protein that inhibits the growth of muscle tissue. MYO-029 is a recombinant human antibody designed to bind and inhibit the activity of myostatin.[56] A 2005/2006 study was completed by Wyeth in Collegeville, PA, and included participants afflicted with FSHD. The study could not prove its efficacy and did not proceed further.
- aTyr Pharma First FSHD Patient Study of ResolarisTM -- First Physiocrine-Based Therapeutic Administered to Patients.[57]
- "The Phase 1b/2 study is a double-blind, placebo-controlled, multiple ascending dose trial in up to 44 FSHD patients at multiple sites in the European Union. The exploratory trial is designed to evaluate safety, tolerability, pharmacokinetics and the biological activity of ResolarisTM in adult patients with FSHD. Our ResolarisTM FSHD trial represents the first patient administration of a naturally occurring protein derived from a new class of physiological modulators, Physiocrines. We believe this trial will be an important step in our plan to develop new medicines that will have a meaningful impact for patients by activating physiological pathways important to skeletal muscle health.[58]
Procedures used to improve quality of life
- Quality of life can be measured with specific questionnaires.[59]
- Scapular fusion: surgical fusion of the scapula to the thorax.
- Scapular bracing: a scapular brace helps stabilize the shoulder and correct glenohumeral positioning.
History
FSHD was first described in 1884 by French physicians Louis Landouzy and Joseph Dejerine. In their paper of 1886, Landouzy and Dejerine drew attention to the familial nature of the disorder and mentioned that four generations were affected in the kindred that they had investigated.[60] Formal definition of FSHD's clinical features didn't occur until 1952 when a large Utah family with FSHD was studied. Beginning about 1980 an increasing interest in FSHD led to increased understanding of the great variability in the disease and a growing understanding of the genetic and pathophysiological complexities. By the late 1990s, researchers were finally beginning to understand the regions of Chromosome 4 associated with FSHD.[10]
Since the publication of the unifying theory in 2010, researchers continued to refine their understanding of DUX4. With increasing confidence in this work, researchers proposed the first a consensus view in 2014 of the pathophysiology of the disease and potential approaches to therapeutic intervention based on that model.[9]
A chronology of important milestones in the history of genetic research related to FSHD is included below in the Genetics section.
Over the years, FSHD has, at various times, been referred to as:
- Landouzy-Dejerine[2]
- Landouzy-Dejerine syndrome[60]
- Erb-Landouzy-Dejerine syndrome[60]
- Landouzy-Dejerine dystrophy or atrophy[60]
- faciohumeroscapular
In Fiction
- In the Amazon Video series The Man in the High Castle, Obergruppenführer John Smith's son, Thomas, is diagnosed with Landouzy-Dejerine syndrome. In the alternate history of the series in which the Axis powers won World War II, Nazi policy is to euthanize those affected by this and other serious conditions to preclude their "drag on society."
References
- ↑ Rickard, Amanda; Petek, Lisa; Miller, Daniel (August 5, 2015). "Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways". Hum. Mol. Genet. 24: 5901–14. PMC 4581613
 . PMID 26246499. doi:10.1093/hmg/ddv315. Retrieved September 10, 2015.
. PMID 26246499. doi:10.1093/hmg/ddv315. Retrieved September 10, 2015. - 1 2 disease overview, MDA, date accessed 6 March 2007
- ↑ Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. (November 2007). "Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy". Am. J. Hum. Genet. 81 (5): 884–94. PMC 2265642 . PMID 17924332. doi:10.1086/521986.
- 1 2 Prevalence of rare diseases: Bibliographic data, www.orpha.net, May 2014, Number 1, Orphanet Report Series
- ↑ Deenen JC, Arnts H, van der Maarel SM, Padberg GW, Verschuuren JJ, Bakker E, Weinreich SS, Verbeek AL, van Engelen BG (2014). "Population-based incidence and prevalence of facioscapulohumeral dystrophy". Neurology. 83 (12): 1056–9. PMC 4166358 . PMID 25122204. doi:10.1212/WNL.0000000000000797.
- ↑ Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW (2004). "Ventilatory support in facioscapulohumeral muscular dystrophy". Neurology. 63 (1): 176–8. PMID 15249635. doi:10.1212/01.wnl.0000133126.86377.e8.
- 1 2 3 Kolata, Gina (19 August 2010). "Reanimated 'Junk' DNA Is Found to Cause Disease". New York Times. Retrieved 29 August 2010.
- 1 2 Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM (19 August 2010). "A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy". Science. 329 (5999): 1650–3. PMID 20724583. doi:10.1126/science.1189044.
- 1 2 3 4 5 6 Tawil, Rabi; van der Maarel, SM; Tapscott, SJ (10 June 2014). "Facioscapulohumeral dystrophy: the path to consensus on pathophysiology". Skeletal Muscle. 4 (1): 12. doi:10.1186/2044-5040-4-12.
- 1 2 3 4 Impossible Things: Through the looking glass with FSH Dystrophy Researchers, Margaret Wahl, MDA, Quest magazine, Vol 14, No 2, March–April 2007
- ↑ Dixit M, Ansseau E, Tassin A, et al. (November 2007). "DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1". Proc. Natl. Acad. Sci. U.S.A. 104 (46): 18157–62. PMC 2084313 . PMID 17984056. doi:10.1073/pnas.0708659104.
- ↑ Rossi M, Ricci E, Colantoni L, et al. (2007). "The Facioscapulohumeral muscular dystrophy region on 4qter and the homologous locus on 10qter evolved independently under different evolutionary pressure". BMC Med. Genet. 8: 8. PMC 1821008 . PMID 17335567. doi:10.1186/1471-2350-8-8.
- ↑ Gilbert JR, Stajich JM, Wall S, et al. (August 1993). "Evidence for heterogeneity in facioscapulohumeral muscular dystrophy (FSHD)". Am. J. Hum. Genet. 53 (2): 401–8. PMC 1682358 . PMID 8328457.
- ↑ Speer MC, Pericak-Vance MA, Stajich JM, et al. (September 1997). "Further exclusion of FSHD1B from the telomeric region of 10q". Neurogenetics. 1 (2): 151–2. PMID 10732819. doi:10.1007/s100480050023.
- ↑ Deak KL, Lemmers RJ, Stajich JM, et al. (February 2007). "Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E-11 and D4Z4". Neurology. 68 (8): 578–82. PMID 17229919. doi:10.1212/01.wnl.0000254991.21818.f3.
- 1 2 Lemmers, RJ; Tawil, R; Petek, LM; et al. (Dec 2012). "Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2". Nature Genetics. 44 (12): 1370–1374. PMC 3671095 . PMID 23143600. doi:10.1038/ng.2454.
- ↑ Landouzy; Dejerine (1884). "De la myopathie atrophique progressive (myopathie héréditaire, débutant dans l’enfance par la face, sans altération du système nerveux)". Comptes rendus de l’Académie des sciences. 98: 53–55.
- ↑ Landouzy; Dejerine (1886). "Contribution à l’étude de la myopathie atrophique progressive (myopathie atrophique progressive, à type scapulo-huméral)". Comptes rendus de la Société de biologie. 38: 478–481.
- ↑ Tyler, Frank; Stephens, FE (April 1950). "Studies in disorders of muscle. II Clinical manifestations and inheritance of facioscapulohumeral dystrophy in a large family". Annals of Internal Medicine. 32: 640–660. doi:10.7326/0003-4819-32-4-640.
- ↑ Padberg, GW (1982-10-13). "Facioscapulohumeral disease (Thesis)". Leiden University.
- ↑ Koenig, M; Hoffman, EP; Bertelson, CJ; Monaco, AP; Feener, C; Kunkel, LM (Jul 31, 1987). "Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals". Cell. 50: 509–517. doi:10.1016/0092-8674(87)90504-6.
- ↑ Wijmenga, C; Padberg, GW; Moerer, P; et al. (April 1991). "Mapping of facioscapulohumeral muscular dystrophy gene to chromosome 4q35-qter by multipoint linkage analysis and in situ hybridization". Genomics. 9: 570–575. doi:10.1016/0888-7543(91)90348-I.
- ↑ Wijmenga, C; Hewitt, JE; Sandkuijl, LA; et al. (Sep 1992). "Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy". Nature Genetics. 2 (1): 26–30. doi:10.1038/ng0992-26.
- ↑ van Deutekom, JC; Wijmenga, C; van Tienhoven, EA; et al. (Dec 1993). "FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit". Human Molecular Genetics. 2 (12): 2037–2042. doi:10.1093/hmg/2.12.2037.
- ↑ Gilbert, JR; Stajich, JM; Wall, S; et al. (Aug 1993). "Evidence for heterogeneity in facioscapulohumeral muscular dystrophy (FSHD)". American Journal of Human Genetics. 53 (2): 401–408. PMC 1682358 . PMID 8328457.
- 1 2 Winokur, ST; Bengtsson, U; Feddersen, J; et al. (May 1994). "The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: implications for a role of chromatin structure in the pathogenesis of the disease". Chromosome Research. 2: 225–234. doi:10.1007/bf01553323.
- ↑ Hewitt, JE; Lyle, R; Clark, LN; et al. (Aug 1994). "Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy". Human Molecular Genetics. 3 (8): 1287–1295. doi:10.1093/hmg/3.8.1287.
- ↑ Gilbert, JR; Speer, MC; Stajich, J; et al. (Oct 1995). "Exclusion mapping of chromosomal regions which cross hybridise to FSHD1A associated markers in FSHD1B". Journal of Medical Genetics. 32 (10): 770–773. doi:10.1136/jmg.32.10.770.
- ↑ van Deutekom, JC; Lemmers, RJ; Grewal, PK; et al. (May 1996). "Identification of the first gene (FRG1) from the FSHD region on human chromosome 4q35". Human Molecular Genetics. 5 (5): 581–590. PMID 8733123. doi:10.1093/hmg/5.5.581.
- ↑ Tupler, R; Barbierato, L; et al. (Sep 1998). "Identical de novo mutation at the D4F104S1 locus in monozygotic male twins affected by facioscapulohumeral muscular dystrophy (FSHD) with different clinical expression". Journal of Medical Genetics. 35 (9): 778–783. doi:10.1136/jmg.35.9.778.
- 1 2 Kowaljow, V; Marcowycz, A; Ansseau, E; et al. (Aug 2007). "The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein". Neruomuscular Disorders. 17: 611–623. doi:10.1016/j.nmd.2007.04.002.
- ↑ Gabriels, J; Beckers, MC; Ding, H; et al. (Aug 5, 1999). "Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element". Gene. 236 (1): 25–32. PMID 10433963. doi:10.1016/S0378-1119(99)00267-X.
- ↑ Tsien, F; Sun, B; Hopkins, NE; et al. (Nov 2001). "Methylation of the FSHD syndrome-linked subtelomeric repeat in normal and FSHD cell cultures and tissues". Molecular Genetics and Metabolism. 74: 322–331. PMID 11708861. doi:10.1006/mgme.2001.3219.
- ↑ Lemmers, RJ; de Kievit, P; Sandkuijl, L; et al. (Oct 2002). "Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere". Nature Genetics. 32 (2): 235–236. PMID 12355084. doi:10.1038/ng999.
- ↑ Gabellini, D; Green, MR; Tupler, R (Aug 9, 2002). "Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle". Cell. 110: 339–348. doi:10.1016/S0092-8674(02)00826-7.
- ↑ van Overveld, PG; Lemmers, RJ; Sandkuijl, LA; et al. (Dec 2003). "Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy". Nature Genetics. 35 (4): 315–317. PMID 14634647. doi:10.1038/ng1262.
- ↑ Lemmers, RJ; Wohlgemuth, M; Frants, RR; Padberg, GW; Morava, E; van der Maarel, SM (Dec 2004). "Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy". The American Journal of Human Genetics. 75: 1124–1130. PMC 1182148 . PMID 15467981. doi:10.1086/426035.
- ↑ Gabellini, D; D'Antona, G; Moggio, M; et al. (Feb 23, 2006). "Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1". Nature. 439 (7079): 973–977. PMID 16341202. doi:10.1038/nature04422.
- ↑ Clapp, J; Mitchell, LM; Bolland, DJ; et al. (Aug 2007). "Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy". The American Journal of Human Genetics. 81: 264–279. doi:10.1086/519311.
- ↑ Dixit, M; Ansseau, E; Tassin, A; et al. (Nov 13, 2007). "DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1". Proceedings of the National Academy of Sciences of the USA. 104 (46): 18157–18162. PMC 2084313 . PMID 17984056. doi:10.1073/pnas.0708659104.
- ↑ de Greef, JC; Lemmers, RJ; van Engelen, BG; et al. (Oct 2009). "Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD". Human Mutation. 30: 1449–1459. PMID 19728363. doi:10.1002/humu.21091.
- ↑ Snider, L; Asawachaicharn, A; Tyler, AE; et al. (Jul 1, 2009). "RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy". Human Molecular Genetics. 18 (13): 2414–2430. PMC 2694690 . PMID 19359275. doi:10.1093/hmg/ddp180.
- ↑ Lemmers, RJ; van der Vliet, PJ; Klooster, R; et al. (Sep 24, 2010). "A unifying genetic model for facioscapulohumeral muscular dystrophy" (PDF). Science. 329 (5999): 1650–1653. PMID 20724583. doi:10.1126/science.1189044.
- ↑ Snider, L; Geng, LN; Lemmers, RJ; et al. (Oct 2010). "Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene". PLoS Genetics. 6: e1001181. PMC 2965761 . PMID 21060811. doi:10.1371/journal.pgen.1001181.
- ↑ Scionti, I; Greco, F; Ricci, G; et al. (Apr 6, 2012). "Large-scale population analysis challenges the current criteria for the molecular diagnosis of fascioscapulohumeral muscular dystrophy". The American Journal of Human Genetics. 90: 628–635. PMC 3322229 . PMID 22482803. doi:10.1016/j.ajhg.2012.02.019.
- ↑ Geng, LN; Yao, Z; Snider, L; et al. (Jan 17, 2012). "DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy". Developmental Cell. 22: 38–51. PMC 3264808 . PMID 22209328. doi:10.1016/j.devcel.2011.11.013.
- ↑ Jones, TI; Chen, JC; Rahimov, F; et al. (Oct 15, 2012). "Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: evidence for disease modifiers and a quantitative model of pathogenesis". Human Molecular Genetics. 21 (20): 4419–4430. PMID 22798623. doi:10.1093/hmg/dds284.
- ↑ Cabianca, DS; Casa, Casa; Bodega, B; et al. (May 11, 2012). "A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy". Cell. 149: 819–831. PMC 3350859 . PMID 22541069. doi:10.1016/j.cell.2012.03.035.
- ↑ Sacconi, S; Lemmers, RJ; Balog, J; et al. (Oct 3, 2013). "The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1". The American Journal of Human Genetics. 93: 744–751. PMC 3791262 . PMID 24075187. doi:10.1016/j.ajhg.2013.08.004.
- ↑ Krom, YD; Thijssen, PE; Young, JM; et al. (Apr 2013). "Intrinsic Epigenetic Regulation of the D4Z4 Macrosatellite Repeat in a Transgenic Mouse Model for FSHD". PLoS Genetics. 9: e1003415. PMC 3616921 . PMID 23593020. doi:10.1371/journal.pgen.1003415.
- ↑ Ferreboeuf, M; Mariot, V; Bessieres, B; et al. (Jan 1, 2014). "DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles" (PDF). Human Molecular Genetics. 23: 171–181. PMID 23966205. doi:10.1093/hmg/ddt409.
- ↑ http://www.fshsociety.org/
- ↑ http://www.fshdglobal.org/
- ↑ http://www.fshd-europe.org/
- ↑ FSHD Fact Sheet, MDA, 11/1/2001
- ↑ Wyeth Initiates Clinical Trial with Investigational Muscular Dystrophy Therapy MYO-029
- ↑ "aTyr Pharma | Brave Science | Meaningful Medicines". www.atyrpharma.com. Retrieved 2015-09-16.
- ↑ "Home - ClinicalTrials.gov". ClinicalTrials.gov. Retrieved 2015-09-16.
- ↑ Dany, Antoine; Barbe, Coralie; Rapin, Amandine; Réveillère, Christian; Hardouin, Jean-Benoit; Morrone, Isabella; Wolak-Thierry, Aurore; Dramé, Moustapha; Calmus, Arnaud; Sacconi, Sabrina; Bassez, Guillaume; Tiffreau, Vincent; Richard, Isabelle; Gallais, Benjamin; Prigent, Hélène; Taiar, Redha; Jolly, Damien; Novella, Jean-Luc; Boyer, François Constant (2015). "Construction of a Quality of Life Questionnaire for slowly progressive neuromuscular disease". Quality of Life Research. 24 (11): 2615–2623. ISSN 0962-9343. doi:10.1007/s11136-015-1013-8.
- 1 2 3 4 Landouzy-Dejerine syndrome, whonamedit.com, date accessed March 11, 2007
External links
- list of clinical trials for FSHD, Clinicaltrials.gov.
- FSH Society
- FSHD Global Research Foundation
- FSHD Champions
- Photo and additional links at Washington University
- Richard Fields Center for FSHD and Neuromuscular Research
- National Registry of Myotonic Dystrophy and Facioscapulohumeral Muscular Dystrophy Patients and Family Members
- The FSH Society
- Friends of FSH Research
- European FSH patients Group & FSH-EUROPE
- The MDA addresses the gamut of issues related to Muscular Dystrophy.
- Muscular Dystrophy Association's website in Greece
- fsh at NIH/UW GeneTests
| Wikimedia Commons has media related to FSHD. |