Primary ciliary dyskinesia
| Primary ciliary dyskinesia | |
|---|---|
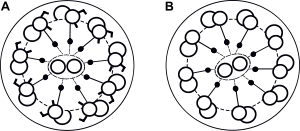 | |
| Normal cilia (A) and cilia representative of Kartagener's syndrome (B). | |
| Classification and external resources | |
| Specialty | pulmonology |
| ICD-10 | J98.0* |
| ICD-9-CM | 759.3* |
| OMIM | 244400 242650 |
| DiseasesDB | 7112 29887 |
| eMedicine | med/1220 ped/1166 |
| MeSH | D002925 |
Primary ciliary dyskinesia (PCD), also called immotile ciliary syndrome or Kartagener syndrome, is a rare, ciliopathic, autosomal recessive genetic disorder that causes defects in the action of cilia lining the respiratory tract (lower and upper, sinuses, Eustachian tube, middle ear), fallopian tube, and flagella of sperm cells. The phrase "immotile ciliary syndrome" is no longer favored as the cilia do have movement, but are merely inefficient or unsynchronized.
Respiratory epithelial motile cilia, which resemble microscopic "hairs" (although structurally and biologically unrelated to hair), are complex organelles that beat synchronously in the respiratory tract, moving mucus toward the throat. Normally, cilia beat 7 to 22 times per second, and any impairment can result in poor mucociliary clearance, with subsequent upper and lower respiratory infection. Cilia also are involved in other biological processes (such as nitric oxide production), which are currently the subject of dozens of research efforts. As the functions of cilia become better understood, the understanding of PCD should be expected to advance.
Classification

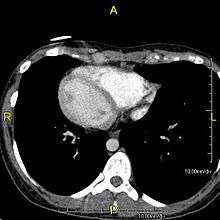
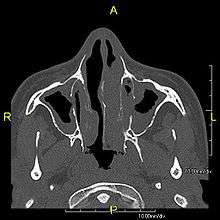
When accompanied by the combination of situs inversus (reversal of the internal organs), chronic sinusitis, and bronchiectasis, it is known as Kartagener syndrome (only 50% of primary ciliary dyskinesia cases include situs inversus).
Signs and symptoms
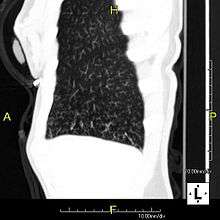
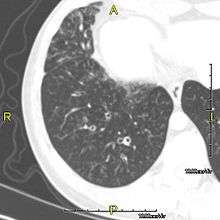
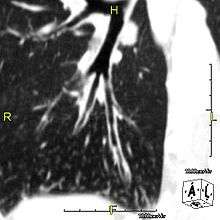
The main consequence of impaired ciliary function is reduced or absent mucus clearance from the lungs, and susceptibility to chronic recurrent respiratory infections, including sinusitis, bronchitis, pneumonia, and otitis media. Progressive damage to the respiratory system is common, including progressive bronchiectasis beginning in early childhood, and sinus disease (sometimes becoming severe in adults). However, diagnosis is often missed early in life despite the characteristic signs and symptoms.[1] In males, immotility of sperm can lead to infertility, although conception remains possible through the use of in vitro fertilization and, as well as this, there have been reported cases where sperm were able to move.[2] Trials have also shown that there is a marked reduction in fertility in female sufferers of Kartagener's Syndrome due to dysfunction of the oviductal cilia.[3]
Many affected individuals experience hearing loss and show symptoms of otitis media which demonstrate variable responsiveness to the insertion of myringotomy tubes or grommets. Some patients have a poor sense of smell, which is believed to accompany high mucus production in the sinuses (although others report normal - or even acute - sensitivity to smell and taste). Clinical progression of the disease is variable, with lung transplantation required in severe cases. Susceptibility to infections can be drastically reduced by an early diagnosis. Treatment with various chest physiotherapy techniques has been observed to reduce the incidence of lung infection and to slow the progression of bronchiectasis dramatically. Aggressive treatment of sinus disease beginning at an early age is believed to slow long-term sinus damage (although this has not yet been adequately documented). Aggressive measures to enhance clearance of mucus, prevent respiratory infections, and treat bacterial superinfections have been observed to slow lung-disease progression. Although the true incidence of the disease is unknown, it is estimated to be 1 in 32,000,[4] although the actual incidence may be as high as 1 in 15,000.
Genetics
PCD is a genetically heterogeneous disorder affecting motile cilia[5] which are made up of approximately 250 proteins.[6] Around 90%[7] of individuals with PCD have ultrastructural defects affecting protein(s) in the outer and/or inner dynein arms which give cilia their motility, with roughly 38%[7] of these defects caused by mutations on two genes, DNAI1 and DNAH5, both of which code for proteins found in the ciliary outer dynein arm.
There is an international effort to identify genes that code for inner dynein arm proteins or proteins from other ciliary structures (radial spokes, central apparatus, etc.) associated with PCD. The role of DNAH5 in heterotaxy syndromes and left-right asymmetry is also under investigation.
| Type | OMIM | Gene | Locus |
|---|---|---|---|
| CILD1 | 244400 | DNAI1 | 9p21-p13 |
| CILD2 | 606763 | ? | 19q13.3-qter |
| CILD3 | 608644 | DNAH5 | 5p |
| CILD4 | 608646 | ? | 15q13 |
| CILD5 | 608647 | ? | 16p12 |
| CILD6 | 610852 | TXNDC3 | 7p14-p13 |
| CILD7 | 611884 | DNAH11 | 7p21 |
| CILD8 | 612274 | ? | 15q24-q25 |
| CILD9 | 612444 | DNAI2 | 17q25 |
| CILD10 | 612518 | KTU | 14q21.3 |
| CILD11 | 612649 | RSPH4A | 6q22 |
| CILD12 | 612650 | RSPH9 | 6p21 |
| CILD13 | 613190 | LRRC50 | 16q24.1 |
Pathophysiology
This condition is genetically inherited. Structures that make up the cilia including inner and/or outer dynein arms, central apparatus, radial spokes, etc. are missing or dysfunctional and thus the axoneme structure lacks the ability to move. Axonemes are the elongated structures that make up cilia and flagella. Additionally, there may be chemical defects that interfere with ciliary function in the presence of adequate structure. Whatever the underlying cause, dysfunction of the cilia begins during and impacts the embryologic phase of development.
Specialised monocilia are at the heart of this problem. They lack the central-pair microtubules of ordinary motile cilia and so rotate clockwise rather than beat; in the primitive knot at the anterior end of the primitive streak in the embryo, these are angled posteriorly[8][9] such that they prescribe a D-shape rather than a circle.[9] This has been shown to generate a net leftward flow in mouse and chick embryos, and sweeps the protein to the left, triggering normal asymmetrical development.
However, in some individuals with PCD, mutations thought to be in the gene coding for the key structural protein left-right dynein (lrd)[5] result in monocilia which do not rotate. There is therefore no flow generated in the node, Shh moves at random within it, and 50% of those affected develop situs inversus which can occur with or without dextrocardia, where the laterality of the internal organs is the mirror-image of normal. Affected individuals therefore have Kartagener syndrome. This is not the case with some PCD-related genetic mutations: at least 6% of the PCD population have a condition called situs ambiguus or heterotaxy where organ placement or development is neither typical (situs solitus) nor totally reversed (situs inversus totalis) but is a hybrid of the two. Splenic abnormalities such as polysplenia, asplenia and complex congenital heart defects are more common in individuals with situs ambiguus and PCD, as they are in all individuals with situs ambiguus.[10]
The genetic forces linking failure of nodal monocilia and situs issues and the relationship of those forces to PCD are the subject of intense research interest. For now hypotheses abound—some, like the one above, are generally accepted. However, knowledge in this area is constantly advancing.
Relation to other rare genetic disorders
Recent findings in genetic research have suggested that a large number of genetic disorders, both genetic syndromes and genetic diseases, that were not previously identified in the medical literature as related, may be, in fact, highly related in the genetypical root cause of the widely varying, phenotypically-observed disorders. Thus, PCD is a ciliopathy. Other known ciliopathies include Bardet-Biedl syndrome, polycystic kidney and liver disease, nephronophthisis, Alstrom syndrome, Meckel-Gruber syndrome and some forms of retinal degeneration.[11]
Diagnosis
Several diagnostic tests for this condition have been proposed.[12] These include nasal nitric oxide levels, light microscopy of biopsies for ciliary beat pattern and frequency and electron microscopic examination of dynein arms. Genetic testing has also been proposed but this is difficult given that there are multiple genes involved.
Treatment
There no standardized effective treatment strategies for the condition. Severe fatal respiratory failure can develop; long-term treatment with macrolides such as clarithromycin, erythromycin and azithromycin has been empirically applied for the treatment of primary ciliary dyskinesia in Japan, though controversial due to the effects of the medications. [13]
Prognosis
A 1998 review noted that life expectancy is usually normal, but that there have occasionally been reported neonatal deaths due to PCD.[14] A 2016 longitudinal study followed 151 adults with PCD for a median of 7 years. Within that span, 7 persons died with a median age of 65.[15]
History
The classic symptom combination associated with PCD was first described by A. K. Zivert[16] in 1904, while Kartagener published his first report on the subject in 1933.[17]
References
- ↑ Coren, Me; Meeks, M; Morrison, I; Buchdahl, Rm; Bush, A (2002). "Primary ciliary dyskinesia: age at diagnosis and symptom history". Acta paediatrica (Oslo, Norway : 1992). 91 (6): 667–9. PMID 12162599. doi:10.1080/080352502760069089.
- ↑ http://www.pcdsupport.org.uk/index.php/faqs/will_it_be_difficult_to_have_children/
- ↑ "The oviductal cilia and Kartagener's syndrome, along with an increase in ectopic pregnancy.". Fertil Steril. 46 (3): 412–6. Sep 1986. PMID 3488922.
- ↑ Ceccaldi PF, Carre-Pigeon F, Youinou Y, Delepine B, Bryckaert PE, Harika G, Quereux C, Gaillard D (2004). "Kartagener's syndrome and infertility: observation, diagnosis and treatment". J Gynecol Obstet Biol Reprod. (Paris). 33 (3): 192–4.
- 1 2 Chodhari, R; Mitchison, HM; Meeks, M (March 2004). "Cilia, primary ciliary dyskinesia and molecular genetics". Paediatric respiratory reviews. 5 (1): 69–76. PMID 15222957. doi:10.1016/j.prrv.2003.09.005.
- ↑ GeneReviews. "Primary Ciliary Dyskinesia". Retrieved 2007-11-16.
- 1 2 Zariwala, MA; Knowles, MR; Omran, H (2007). "Genetic defects in ciliary structure and function". Annual Review of Physiology. 69 (1): 423–50. PMID 17059358. doi:10.1146/annurev.physiol.69.040705.141301.
- ↑ Cartwright, Jh; Piro, O; Tuval, I (May 2004). "Fluid-dynamical basis of the embryonic development of left-right asymmetry in vertebrates" (Free full text). Proceedings of the National Academy of Sciences of the United States of America. 101 (19): 7234–9. PMC 409902
 . PMID 15118088. doi:10.1073/pnas.0402001101.
. PMID 15118088. doi:10.1073/pnas.0402001101. - 1 2 Nonaka, S; Yoshiba, S; Watanabe, D; Ikeuchi, S; Goto, T; Marshall, Wf; Hamada, H (August 2005). "De novo formation of left-right asymmetry by posterior tilt of nodal cilia" (Free full text). PLoS Biology. 3 (8): e268. PMC 1180513 . PMID 16035921. doi:10.1371/journal.pbio.0030268.

- ↑ Kennedy, Mp; Omran, H; Leigh, Mw; Dell, S; Morgan, L; Molina, Pl; Robinson, Bv; Minnix, Sl; Olbrich, H; Severin, T; Ahrens, P; Lange, L; Morillas, Hn; Noone, Pg; Zariwala, Ma; Knowles, Mr (June 2007). "Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia" (Free full text). Circulation. 115 (22): 2814–21. PMID 17515466. doi:10.1161/CIRCULATIONAHA.106.649038.
- ↑ Badano, Jose L.; Norimasa Mitsuma; Phil L. Beales; Nicholas Katsanis (September 2006). "The Ciliopathies : An Emerging Class of Human Genetic Disorders". Annual Review of Genomics and Human Genetics. 7 (1): 125–148. PMID 16722803. doi:10.1146/annurev.genom.7.080505.115610. Retrieved 2008-06-15.
- ↑ Shoemark A, Moya E, Hirst RA, Patel MP, Robson EA, Hayward J, Scully J, Fassad MR, Lamb W, Schmidts M, Dixon M, Patel-King RS, Rogers AV,, Rutman A, Jackson CL, Goggin P, Rubbo B, Ollosson S, Carr S, Walker W, Adler B, Loebinger MR, Wilson R, Bush A, Williams H, Boustred C, Jenkins L, Sheridan E, Chung EMK, Watson CM, Cullup T, Lucas JS, Kenia P, O'Callaghan C, King SM, Hogg C, Mitchison HM (2017) High prevalence of CCDC103 p.His154Pro mutation causing primary ciliary dyskinesia disrupts protein oligomerisation and is associated with normal diagnostic investigations. Thorax pii: thoraxjnl-2017-209999. doi: 10.1136/thoraxjnl-2017-209999
- ↑ Kido, Takashi ; et al. (2012). "Two Cases of Primary Ciliary Dyskinesia with Different Responses to Macrolide Treatment". Internal Medicine. 51 (9): 1093–1098. ISSN 1349-7235. doi:10.2169/internalmedicine.51.6617. Retrieved May 18, 2017.
- ↑ Bush, A.; Cole, P.; Hariri, M.; Mackay, I.; Phillips, G.; O'Callaghan, C.; Wilson, R.; Warner, J. O. (1998-10-01). "Primary ciliary dyskinesia: diagnosis and standards of care". European Respiratory Journal. 12 (4): 982–988. ISSN 0903-1936. PMID 9817179. doi:10.1183/09031936.98.12040982.
- ↑ Shah, Anand; Shoemark, Amelia; MacNeill, Stephanie J.; Bhaludin, Basrull; Rogers, Andrew; Bilton, Diana; Hansell, David M.; Wilson, Robert; Loebinger, Michael R. (2016-08-01). "A longitudinal study characterising a large adult primary ciliary dyskinesia population". European Respiratory Journal. 48 (2): 441–450. ISSN 0903-1936. PMID 27288033. doi:10.1183/13993003.00209-2016.
- ↑ Zivert AK (1904). "Über einen Fall von Bronchiectasie bei einem Patienten mit situs inversus viscerum". Berliner klinische Wochenschrift. 41: 139–141.
- ↑ Kartagener M (1933). "Zur Pathogenese der Bronchiektasien: Bronchiektasien bei Situs viscerum inversus". Beiträge zur Klinik der Tuberkulose. 83 (4): 489–501. doi:10.1007/BF02141468.
External links
- GeneReview/NCBI/NIH/UW entry on Primary Ciliary Dyskinesia
- http://www.pcdfoundation.org
- http://www.pcdsupport.org.uk
- http://www.med.unc.edu/cystfib/PCD.htm
- http://www.cheo.on.ca/english/9301c.shtml
- http://www.pcdforum.com/
- Kartagener's syndrome on Who Named It
- http://micro.magnet.fsu.edu/cells/ciliaandflagella/ciliaandflagella.html Cilia defined
- http://micro.magnet.fsu.edu/cells/ciliaandflagella/ciliaandflagella.html Cilia defined
- http://www.dyskinesia.be Belgian Association about Primary Ciliary Dyskinesia
This article may contain some text from the public domain source "National Heart, Lung, and Blood Institute Rare Diseases Report FY 2001" available at http://www.nhlbi.nih.gov/resources/docs/raredisrpt01.htm