Intramolecular aglycon delivery
Intramolecular aglycon delivery is a synthetic strategy for the construction of glycans. This methodology is generally applied to the formation of difficult glycosidic linkage.
Introduction
Glycosylation reactions are very important reactions in carbohydrate chemistry leading to the synthesis of oligosaccharides, preferably in a stereoselective manner. The stereoselectivity of these reactions has been shown to be affected by both the nature and the configuration of the protecting group at C-2 on the glycosyl donor ring. While 1,2-trans-glycosides (e.g. alpha-mannosides and β-glucosides) can be synthesised easily in the presence of a participating group (such as OAc, or NHAc) at the C-2 position in the glycosyl donor ring, 1,2-cis-glycosides are more difficult to prepare. 1,2-cis-glycosides with the alpha configuration (e.g. glucosides or galactosides) can often be prepared using a non-participating protecting group (such as Bn, or All) at C-2 (OH). However, 1,2-cis-glycosides with the β configuration are the most difficult to achieve, and present the greatest challenge in glycosylation reactions.

One of the most recent approaches to prepare 1,2-cis-β-glycosides in a stereospecific manner is termed ‘Intramolecular Aglycon Delivery’, and various methods have been developed under this approach.[1] In this methodology, the glycosyl acceptor is tethered into the C-2-O-protecting group (X) in the first step. Upon activation of the glycosyl donor group (Y) (usually SR, OAc, or Br group) in the next step, the tethered aglycon traps the developing oxocarbenium ion at C-1 and is transferred from the same face as OH-2, forming the glycosidic bond stereospecifically.
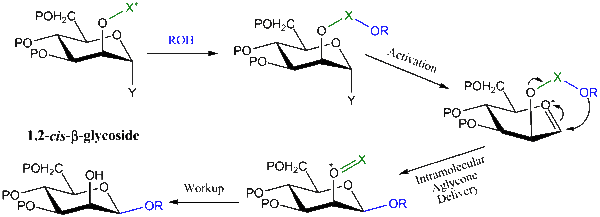
The yield of this reaction drops as the bulkiness of the alcohol increases.
Intramolecular Aglycon Delivery (IAD) methods
Carbon tethering
Acid catalysed tethering on enol ethers
In this method, the glycosyl donor is protected at the C-2 position by an OAc group. The C-2-OAc protecting group is transformed into an enol ether by the Tebbe reagent (Cp2Ti=CH2), and then the glycosyl acceptor is tethered to the enol ether under acid catalysed conditions to generate a mixed acetal. In a subsequent step, the β-mannoside is formed upon activation of the (Y) group followed by work up.[2]

Iodonium tethering on enol ethers
This method is similar to the previous method in that the glycosyl donor is protected at C-2 by an OAc group, which is converted into an enol ether by the Tebbe reagent. However, in this approach, N-iodosuccinimide (NIS) is used to tether the glycosyl acceptor to the enol ether, and in a second step, activation of the anomeric leaving group leads to intramolecular delivery of the aglycon to C-1 and formation of the 1,2-cis glycoside product.[3]

Iodonium tethering on prop-1-enyl ethers
The glycosyl donor is protected at C-2 by OAll group. The allyl group is then isomerized to prop-1-enyl ether using a Rhodium hydride generated from Wilkinson's catalyst ((PPh3)3RhCl) and butyl lithium. The resulting enol ether is then reacted with NIS and the glycosyl acceptor to generate a mixed acetal. The 1,2-cis (e.g. β-mannosyl) product is formed in the last step upon activation of the anomeric leaving group, delivery of the aglycon from the mixed acetal and finally hydrolytic work-up to remove the remains of the propenyl ether from O-2.[4]

Oxidative tethering on para-methoxybenzyl (PMB) ethers
In this method, the glycosyl donor is protected at C-2 by para-methoxy benzyl (PMB) group. The glycosyl acceptor is then tethered at the benzylic position of the PMB protecting group in the presence of dichloro dicyano quinone (DDQ). The anomeric leaving group (Y) is then activated, and the developing oxocarbenium ion is captured by the tethered aglycon alcohol (OR) to give 1,2-cis-β glycoside product.[5]

Solid-supported oxidative tethering on para-alkoxybenzyl ethers
This is a modification of the method of oxidative tethering to a para-methoxybenzyl ether. The difference here is that the para-alkoxybenzyl group is attached to a solid support; the β-mannoside product is released into the solution phase in the last step, while the by-products remain attached to the solid phase. This makes the purification of the β-glycoside easier; it is formed as the almost exclusive product.[6]

Silicon tethering
The initial step in this method involves the formation of a silyl ether at C-2 (OH) of the glycosyl donor upon addition of dimethyl dichloro silyl in the presence of a strong base such as (BuLi), then the glycosyl acceptor is added to form mixed silaketal. Activation of the anomeric leaving group then in the presence of a hindered base leads to the β-glycoside.[7]

A modified silicon tethering method involves mixing of the glycosyl donor with the glycosyl acceptor and dimethyl dichloro silane in the presence of imidazole to give the mixed silaketal in one pot. Activation of the tethered intermediate then leads to the β-glycoside product.[8]

See also
References
- ↑ Ian Cumpstey. Carbohydrate research, 343 (2008) 1553–1573
- ↑ Barresi, F.; Hindsgaul, O. J. Am. Chem. Soc 1991, 113, 9376–9377
- ↑ Ennis, S. C.; Fairbanks, A. J.; Slinn, C. A.; Tennant-Eyles, R. J.; Yeates, H. S. Tetrahedron 2001, 57, 4221–4230
- ↑ Seward, C. M. P.; Cumpstey, I.; Aloui, M.; Ennis, S. C.; Redgrave, A. J.; Fairbanks, A. J. Chem. Commun. 2000, 1409–1410
- ↑ Ito, Y.; Ogawa, T. Angew. Chem. Int. Ed. Engl. 1994, 33, 1765–1767
- ↑ Ito, Y.; Ogawa, T. J. Am. Chem. Soc. 1997, 119, 5562–5566
- ↑ Stork, G.; Kim, G. J. Am. Chem. Soc. 1992, 114, 1087-1088
- ↑ Stork, G.; La Clair, J. J. J. Am. Chem. Soc. 1996, 118, 247–248