Hydrogen bond catalysis

Hydrogen bond catalysis is a type of organocatalysis that relies on use of hydrogen bonding interactions to accelerate and control organic reactions. In biological systems, hydrogen bonding plays a key role in many enzymatic reactions, both in orienting the substrate molecules and lowering barriers to reaction.[1] However, chemists have only recently attempted to harness the power of using hydrogen bonds to perform catalysis, and the field is relatively undeveloped compared to research in Lewis acid catalysis.[2]
Catalytic amounts of hydrogen bond donors can promote reactions through a variety of different mechanisms. During the course of a reaction, hydrogen bonding can be used to stabilize anionic intermediates and transition states. Alternatively, some catalysts can bind small anions, enabling the formation of reactive electrophilic cations. More acidic donors can act as general or specific acids, which activate electrophiles by protonation. A powerful approach is the simultaneous activation of both partners in a reaction, e.g. nucleophile and electrophile, termed "bifunctional catalysis". In all cases, the close association of the catalyst molecule to substrate also makes hydrogen bond catalysis a powerful method of inducing enantioselectivity.
Hydrogen bonding catalysts are often simple to make, relatively robust, and can be synthesized in high enantiomeric purity. New reactions catalyzed by hydrogen bond donors are being discovered at an increasing pace, including asymmetric variants of common organic reactions useful for synthesis, such as aldol additions, Diels-Alder cycloadditions and Mannich reactions.[3]
However, there are several challenges that must be overcome before hydrogen bond catalysis can achieve its full potential in terms of synthetic utility. Current known reactions are very substrate specific and generally exhibit low rate acceleration and turnover, thus requiring high catalyst loading. Catalysts are often discovered and optimized by trial and error, and chemists have a poor understanding of the relationship between catalyst structure and reactivity. Additionally, the field suffers from a lack of general mechanistic understanding, which has been greatly outpaced by the discovery of new reactions. With more detailed studies of structure and mechanism in the future, hydrogen bond catalysis has great potential for enabling new, efficient, selective reactions and promising applications in asymmetric synthesis.
Catalytic strategies
Stabilization of tetrahedral intermediates
Many useful organic reactions involve the formation of tetrahedral intermediates through nucleophilic attack of functional groups such as aldehydes, amides or imines. In these cases, catalysis with hydrogen bond donors is an attractive strategy since the anionic tetrahedral intermediates are better hydrogen bond acceptors than the starting compound. This means that relative to the initial catalyst-substrate complex, the transition state, bearing more negative charge, is stabilized.
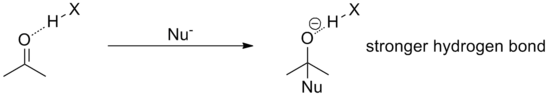
For example, in a typical acyl substitution reaction, the starting carbonyl compound is coordinated to the catalyst through one, two or possibly more hydrogen bonds. During the attack of the nucleophile, negative charge builds on the oxygen until the tetrahedral intermediate is reached. Therefore, the formally negative oxygen engages in a much stronger hydrogen bond than the starting carbonyl oxygen because of its increased negative charge. Energetically, this has the effect of lowering the intermediate and the transition state, thus accelerating the reaction.
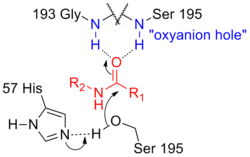
This mode of catalysis is found in the active sites of many enzymes, such as the serine proteases.[4] In this example, the amide carbonyl is coordinated to two N-H donors. These sites of multiple coordination designed to promote carbonyl reactions in biology are termed “oxyanion holes”. Delivery of serine nucleophile forms a tetrahedral intermediate, which is stabilized by the increase hydrogen bonding to the oxyanion hole.
Many synthetic catalysts have been able to successfully employ this strategy to activate a variety of electrophiles. Using a chiral BINOL catalyst, for instance, the Morita-Baylis-Hillman reaction involving the addition of enones to aldehydes can be effected with high enantioselectivity.[5] The nucleophile is an enolate-type species generated from the conjugate addition of PEt3 to the enone, and adds enantioselectively to the aldehyde coordinated to catalyst.

In addition to carbonyls, other electrophiles such as imines can be successfully used. For example, using a simple chiral thiourea catalyst, the asymmetric Mannich reaction of aromatic imines with silyl ketene acetals can be catalyzed with high ee in near quantitative conversion.[6] The mechanism of this reaction is not fully resolved and the reaction is very substrate-specific, only effective on certain aromatic electrophiles.
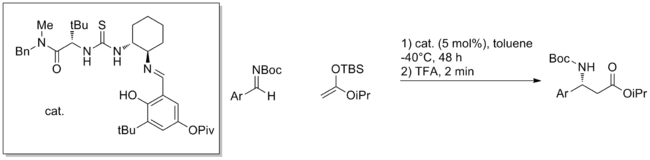
The scope of this mode of activation is immense, with constant new reports of different combinations of electrophiles, nucleophiles and catalyst structures. Furthermore, analogous reactions involving oxyanion intermediates such as enolate addition to nitroso compounds[7] or opening of epoxides[8] have also been successfully catalyzed with this strategy.

However, despite the number of different reactions known, general understanding of the mode of catalysis is limited, and almost all reactions discovered are extremely substrate specific.
Stabilization of anionic fragments
Another strategy that has been explored is the stabilization of reactions that develop partial negative charges in the transition state. Examples of successful applications are most commonly reactions that are approximated concerted and pericyclic in nature. During the course of the reaction, one fragment develops partial negative character and the transition state can be stabilized by accepting hydrogen bond(s).
A demonstrative example is the catalysis of Claisen rearrangements of ester-substituted allyl vinyl ethers reported by the Jacobsen research group.[9] A chiral guanidinium catalyst was found to successfully promote the reaction near room temperature with high enantioselectivity. During the transition state, the fragment coordinated to the amidinium catalyst develops partial anionic character due to the electronegativity of the oxygen and the electron-withdrawing ester group. This increases the strength of hydrogen bonding and lowers the transition state energy, thus accelerating the reaction.

Similarly, negative charge can develop in cycloaddition reactions such as the Diels-Alder reaction, when the partners are appropriately substituted. As a representative example, Rawal and coworkers developed a chiral catalyst based on α,α,α,α-tetraaryl-1,3-dioxolane-4,5-dimethanol (TADDOL) that could catalyze Diels-Alder reactions. In the following example, the reaction with a highly electron-rich diene and an electron-poor dienophile is thought to develop significant negative charge on the enal fragment, and is the transition state is stabilized by increased hydrogen bonding to the TADDOL (Ar = 1-naphthyl).[10]

Anion binding
Hydrogen bond catalysts can also accelerate reactions by assisting in the formation of electrophilic species through abstracting and/or coordinating an anion such as a halide. Urea and thiourea catalysts are the most common donors in anion-binding catalysis, and their ability to bind halides and other anions has been well established in the literature.[11] The use of chiral anion-binding catalysts can create an asymmetric ion pair and induce remarkable stereoselectivity.
One of the first reactions proposed to proceed through anion-binding catalysis is the Pictet-Spengler-type cyclization of hydroxylactams with TMSCl under thiourea catalysis.[12] In the proposed mechanism, after initial substitution of the hydroxyl group with chloride, the key ion pair is formed. The activated iminium ion is closely associated with the chiral thiourea-bound chloride, and intramolecular cyclization proceeds with high stereoselectivity.

Asymmetric ion pairs can also be attacked in intermolecular reactions. In an interesting example, asymmetric addition of enol silane nucleophiles to oxocarbenium ions can be effected by catalytically forming the oxocarbenium through anion binding.[13] Starting from an acetal, the chloroether is generated with boron trichloride and reacted with the enol silane and catalyst. The mechanism of formation of the oxocarbenium-thiourea-chloride complex is not fully resolved. It is thought that under the reaction conditions, the chloroether can epimerize and thiourea can stereoselectively bind chloride to form a closely associated ion pair. This asymmetric ion pair is then attacked by the silane to generate alkylated product.
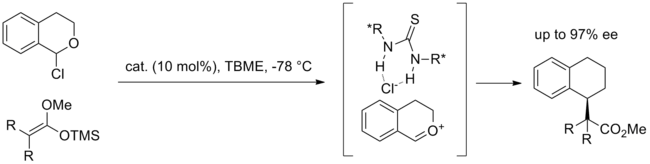
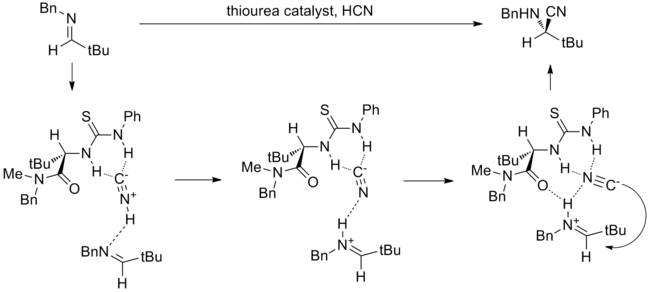
A notable example of the anion-binding mechanism is the hydrocyanation of imines catalyzed by Jacobsen’s amido-thiourea catalyst depicted in the below diagram. This reaction is also one of the most extensively studied through computational, spectroscopic, labeling and kinetic experiments.[14] While direct addition of cyanide to a catalyst-bound imine was considered, an alternative mechanism involving formation of an iminium-cyanide ion pair controlled by catalyst was calculated to have a barrier that is lower by 20 kcal/mol. The proposed most likely mechanism begins with binding of the catalyst to HNC, which exists in equilibrium with HCN. This complex then protonates a molecule of imine, forming an iminium-cyanide ion pair with the catalyst binding and stabilizing the cyanide anion. The iminium is thought to also interact with the amide carbonyl on the catalyst molecule (see bifunctional catalysis below). The bound cyanide anion then rotates, and attacks the iminium through carbon. The investigators conclude that though imine-urea binding was observed through spectroscopy and was supported by early kinetic experiments, imine binding is off-cycle and all evidence points toward this mechanism involving thiourea-bound cyanide.
Protonation
It is often difficult to distinguish between hydrogen-bond catalysis and general acid catalysis.[3] Hydrogen bond donors can have varying acidity, from mild to essentially strong Brønsted acids like phosphoric acids. Looking at the extent of proton transfer over the course of the reaction is challenging and has not been investigated thoroughly in most reactions. Nevertheless, strong acid catalysts are often grouped with hydrogen bond catalysts as they represent on extreme on this continuum and their catalytic behaviors share similarities. The mechanism of activation for these reactions involves initial protonation of the electrophilic partner. This has the effect of rendering the substrate more electrophilic and creating an ion pair, through which it is possible to transfer stereochemical information.

Asymmetric catalysis involving nearly complete protonation of substrate has been effective in Mannich reactions of aromatic aldimines with carbon nucleophiles.[15] In addition, aza-Friedel-Crafts reactions of furans, amidoalkylations of diazocarbonyl compounds, asymmetric hydrophosphonylation of aldimines and transfer hydrogenations have also been reported.[3] Chiral Brønsted acids are often easily prepared from chiral alcohols such as BINOLs, and many are already present in the literature due to their established utility in molecular recognition research.[16]
Multifunctional strategies
One of the main advantages of hydrogen bond catalysis is the ability to construct catalysts that engage in multiple non-covalent interactions to promote the reaction. In addition to using hydrogen bond donors to activate or stabilize a reactive center during the reaction, it is possible to introduce other functional groups, such as Lewis bases, arenes, or addition hydrogen bonding sites to lend additional stabilization or to influence the other reactive partner.
For instance, the natural enzyme chorismate mutase, which catalyzes the Claisen rearrangement of chorismate, features many other interactions in addition to the hydrogen bonds involved in stabilizing the enolate-like fragment, which is an example of the anionic fragment stabilization strategy discussed above.[17] A key interaction is the stabilization of the other cationic allyl fragment through a cation-pi interaction in the transition state. The use of many additional hydrogen bonds has several putative purposes. The stabilization of multiple hydrogen bonds to the enzyme helps overcome the entropic cost of binding. Additionally, the interactions help hold the substrate in a reactive conformation, and the enzyme-catalyzed reaction has near-zero entropy of activation, while typical Claisen rearrangements in solution have very negative entropies of activation.

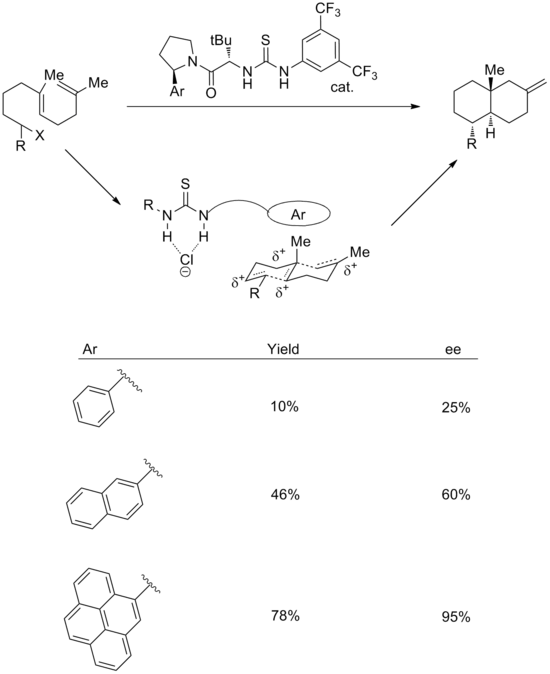
The use of cation-pi interactions has also been successfully implemented in reactions with synthetic catalysts. A combination of anion-binding and cation-pi strategies can be used to effect enantioselective cationic polycyclizations.[18] In the transition state, it is proposed that the thiourea group binds chloride, while the aromatic system stabilizes the associated polyene cation. In support of this, increasing the size of the aromatic ring leads to improvements both in yield and stereoselectivity. The enantioselectivity correlates well with both the polarizability and the quadrupole moment of the aryl group.
Since such a large number of catalysts and reactions involve binding to electrophiles to stabilize the transition state, many bifunctional catalysts also present a Lewis-basic, hydrogen bond acceptor site. As a representative example, Deng and coworkers have developed a thiourea-amine catalyst capable of promoting stereoselective Michael reactions.[19] In the proposed transition state, one of the thiourea N-H donors is coordinated to the Michael acceptor and will stabilize the negative charge buildup. The basic nitrogen lone pair acts as a hydrogen bond acceptor to coordinate the nucleophile, but in the transition state acts as a general base to promote the nucleophilic enolate addition.

This motif of engaging both the nucleophilic and electrophilic partners in a reaction and stabilizing them in the transition state is very common in bifunctional catalysis and many more examples can be found in the article on thiourea organocatalysis.
A relatively new strategy of using synthetic oligopeptides to perform catalysis has yielded many successful examples of catalytic methods.[20] Peptides feature multiple potential sites for hydrogen bonding and it is generally not understood how these engage substrate or how they promote reaction. Peptides have the advantage of being extremely modular and often these catalysts are screened in large arrays. Highly enantioselective reactions have been discovered in this manner such as the aldol reaction depicted below.

Other transformations successfully catalyzed by synthetic peptides include hydrocyanation, acylation, conjugate additions, aldehyde-imine couplings, aldol reaction and bromination. Although the nature of the transition states is unclear, in many examples small changes in the catalyst structure have dramatic effects on reactivity. It is hypothesized that a large number of hydrogen bonds both within the peptide and between catalyst and substrate must cooperate to meet the geometrical requirements for successful catalysis. Beyond this, understanding of catalyst design and mechanism has not yet progressed beyond requiring the testing of libraries of peptides.
Catalyst design
Privileged structures
The types of hydrogen bond donors used in catalysis vary widely from reaction to reaction, even among similar catalytic strategies. While specific systems are often studied and optimized extensively, a general understanding of the optimal donor for a reaction or the relationship between catalyst structure and reactivity is greatly lacking. It is not yet practical to rationally design structures to promote a desired reaction with the desired selectivity. However, contemporary hydrogen bond catalysis is primarily focused on a few types of systems that experimentally seem to be effective in a variety of situations.[21] These are termed “privileged structures”.

- Ureas and thioureas are by far the most common structures and can stabilize a variety of negatively charged intermediates, as well as engage in anion-binding catalysis. Bifunctional urea and thiourea catalysis are abundant in the literature.
- Guanidinium and amidinium ions are structural relatives of ureas and thioureas and can catalyze similar reactions but, by virtue of their positive charge, are stronger donors and much more acidic. The mechanism of guanidinium and amidinium catalysis is thought to often involve partial protonation of substrate.
- Diol catalysts are thought to engage substrate with a single hydrogen bond, with the other hydroxyl participating in an internal hydrogen bond. These are some of the earliest hydrogen bond catalysts investigated. They are most commonly used in stabilizing partial anionic charge in transition states, for example coordinating to aldehyde dienophiles in hetero-Diels-Alder reactions.
- Phosphoric acid catalysts are the most common strong acid catalysts and work by formation of chiral ion pairs with basic substrates such as imines.
Catalyst tuning
In general, acidity of donor sites correlates well with the strength of the donor. For example, it is a common strategy to add electron-withdrawing aryl substituents on a thiourea catalyst, which can increase its acidity and thus the strength of its hydrogen bonding. However, it is still unclear how donor strength correlates with desired reactivity. Importantly, more acidic catalysts are not necessarily more effective. For instance, ureas are less acidic than thioureas by roughly 6 pKa units, but it is not generally true that ureas are significantly worse are catalyzing reactions.[22]
Furthermore, the effect of varying substituents on the catalyst is rarely well understood. Small substituent changes can completely change reactivity or selectivity. An example of this was in the optimization studies of a bifunctional Strecker reaction catalyst, one of the first well-studied thiourea catalysts.[23] Specifically, varying the X substituent on the salicylaldimine substituent, it was found that typical electron-withdrawing or electron-donating substituents had little effect on the rate, but ester substituents such as acetate or pivaloate seemed to cause noticeable rate acceleration. This observation is difficult to rationalize given that the X group is far from the reactive center during the course of the reaction and electronics do not seem to be the cause. In general, despite the relative ease of electronic tuning with organic catalysts, chemists have not yet reached a useful understanding of these modifications.

Synthetic applications
Natural product synthesis
To date, there have been few examples of hydrogen bond catalysis in the synthesis of natural products despite the large number of reactions being discovered. Generally, with high required catalyst loading and often extreme substrate specificity, hydrogen bond catalysis is not yet developed enough to provide useful, general reactions that represent a significant improvement over traditional methods. In the published examples, hydrogen bond catalysis is mainly used in the beginning stages to quickly access early intermediates with high enantiomeric enrichment.
In the Jacobsen synthesis of (+)-yohimbine,[24] an indole alkaloid, an early enantioselective Pictet-Spengler reaction using a pyrrole-substituted thiourea catalyst produced gram-scale quantities of product in 94% ee and 81% yield. The remainder of the synthesis was short, using a reductive amination and an intramolecular Diels-Alder reaction.

In 2008, Takemoto disclosed a concise synthesis of (−)-epibatidine that relied on a Michael cascade, catalyzed by a bifunctional catalyst.[25] After initial asymmetric Michael addition to the β-nitrostyrene, intramolecular Michael addition furnishes the cyclic ketoester product in 75% ee. Standard functional group manipulations and an intramolecular cyclization yields the natural product.

Scalable synthesis of building blocks
Aside from total synthesis, a potentially useful application of hydrogen bond catalysis is the bulk synthesis of difficult-to-access chiral small molecules. A notable example is the gram-scale Strecker synthesis of unnatural amino acids using thiourea catalysis, reported in the journal Nature in 2009.[26] The catalyst, whether polymer-bound or homogeneous, is derived from natural tert-leucine and can catalyze (4 mol% catalyst loading) the formation of the Strecker product from benzhydryl amines and aqueous HCN. Hydrolysis of the nitrile and deprotections produces pure unnatural tert-leucine in 84% overall yield and 99% ee.

Challenges and future goals
Despite the widespread interest in organocatalysis and the large number of new catalytic systems that are continuously being discovered, progress in the understanding of mechanism and catalyst design in the field of hydrogen bond catalysis is extremely limited. Compared to a more developed field like palladium-catalyzed coupling reactions, hydrogen bond catalysis presents many challenges that have not yet been successfully tackled.
- Turnover: While palladium-catalyzed reactions can often be effective with catalyst loading of less than 0.1 mol%, hydrogen bond catalysts are often added in more than 10 mol%. Poor rate acceleration is a general trend that will have to be overcome in order for hydrogen bond catalysis to be a practical synthetic strategy.
- Mechanism: In the future, further investigation of the precise steps involved in the mechanism of hydrogen bond catalysis will be required that will enable chemists to rationally design catalytic strategies for more complex or more useful transformations. For comparison, the basic steps of palladium-catalyzed cross coupling have been systematically and thoroughly studied over the last few decades and have led to dramatic advances in catalytic scope, control and reaction design principles. For example, improved understanding of oxidative addition has led to aryl chlorides becoming practical coupling partners, while improved understanding of reductive elimination has led to the development of new reactions involving sp3 centers. Knowing these fundamental catalytic steps, the ability to rationally plan new reactions and cascades has been extremely useful in the field of total synthesis. In contrast, we lack a general, systematic mechanistic understanding of the steps of hydrogen bond catalysis and how to influence them. Detailed mechanistic studies have so far been limited to individual systems, and their findings have not been of demonstrable predictive use.
- Catalyst: A related challenge is the investigation of how changes in catalyst, structural, conformational, and electronic can be used to rationally influence the reaction. The goal would be to fully understand how to use multiple co-operative interactions to best accelerate a reaction and impart selectivity. Ideally, rational catalyst design will eventually replace screening of families of catalysts and the choice of building blocks will become more systematic.
- Scope: While new reactions are constantly being discovered, most reactions have extremely narrow substrate scope and the reason for such narrow scope is often not understood. In the field of palladium catalysis, after the foundations of mechanistic understanding were established, the scope of reactions saw rapid growth. Knowing the factors that affected each step of catalysis allowed for the chemists to envision and pursue new reactions of high synthetic utility, such as C-H bond activation reactions. In the field hydrogen bond catalysis, chemists have not yet reached a stage where new types of reactivity can be easily and systematically targeted. At this point, reaction discovery is useful, but more detailed mechanistic study is required to realize the full potential of hydrogen bond catalysis.
See also
Further reading
- Hydrogen Bond Catalysis. Evans Group Meeting Presentation by Peter H. Fuller. Link
- Asymmetric Hydrogen Bond Catalysis. MacMillan Group Meeting Presentation by Anthony Mastracchio. Link
- Hydrogen Bonding in Asymmetric Catalysis. Leighton Group Meeting Presentation by Uttam Tambar. Link
- Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Wipf Group Meeting Presentation by Zhenglai Fang Link
- Enantioselective Organocatalysis. Ed. Peter I. Dalko, Wiley-VCH: Weinheim, 2007.
References
- ↑ Jacobsen, E. N.; Knowles, R. R. (September 2010). "Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts". Proc. Natl. Acad. Sci. 107 (48): 20678–20685. PMC 2996434
 . PMID 20956302. doi:10.1073/pnas.1006402107.
. PMID 20956302. doi:10.1073/pnas.1006402107. - ↑ Jacobsen, E. N.; Taylor, M. S. (February 2006). "Asymmetric catalysis by chiral hydrogen-bond donors". Angew. Chem. Int. Ed. 45 (10): 1521–1539. doi:10.1002/anie/200503132.
- 1 2 3 Doyle, Abigail G.; Jacobsen, E. N. (December 2007). "Small-molecule H-bond donors in asymmetric catalysis". Chem. Rev. 107 (12): 5713–5743. PMID 18072808. doi:10.1021/cr068373r.
- ↑ Sinnott, M. (1998). Comprehensive Biological Catalysis, Vol. 1. London: Academic Press. pp. 345–379.
- ↑ McDougal, N. T.; Shaus, S. E. (September 2003). "Asymmetric Morita−Baylis−Hillman reactions catalyzed by chiral Brønsted acids". J. Am. Chem. Soc. 125 (40): 12094–12095. PMID 14518986. doi:10.1021/ja037705w.
- ↑ Wenzel, A. G.; Jacobsen, E. N. (2002). "Asymmetric catalytic Mannich reactions catalyzed by urea derivatives: enantioselective synthesis of β-aryl-β-amino acids". J. Am. Chem. Soc. 124 (44): 12964–12965. doi:10.1021/ja028353g.
- ↑ Yamamoto, H.; Momiyama, N. (September 2004). "Bronsted acid catalysis of achiral enamines for regio- and enantioselective nitroso aldol Synthesis". J. Am. Chem. Soc. 127 (4): 1080–1081. PMC 1460970 . PMID 15669829. doi:10.1021/ja0444637.
- ↑ Hine, J.; Linden, S. M.; Kanagasabapathy, V. M. (December 1985). "Double-hydrogen-bonding catalysis of the reaction of phenyl glycidyl ether with diethylamine by 1,8-biphenylenediol". J. Org. Chem. 50 (25): 5096–5099. doi:10.1021/jo00225a021.
- ↑ Uyeda, C.; Jacobsen, E. N. (July 2008). "Enantioselective Claisen rearrangements with a hydrogen-bond donor catalyst". J. Am. Chem. Soc. 130 (29): 9228–9229. PMC 2547484 . PMID 18576616. doi:10.1021/ja803370x.
- ↑ Rawal, Viresh H.; Thadani, A.N.; Stankovich, A.R. (2004). "Enantioselective Diels-Alder reactions catalyzed by hydrogen bonding". PNAS. 101 (16): 5846–5850. PMC 395998 . PMID 15069185. doi:10.1073/pnas.0308545101.
- ↑ Schmidtchen, F. P.; Berger, M. (August 1997). "Artificial organic host molecules for anions". Chem. Rev. 97 (5): 1609–1646. doi:10.1021/cr9603845.
- ↑ Raheem, I. T.; Thiara, P. S.; Peterson, E. A.; Jacobsen, E. N. (August 2007). "Enantioselective Pictet-Spengler-type Cyclizations of Hydroxylactams: H-Bond Donor Catalysis by Anion Binding". J. Am. Chem. Soc. 129 (44): 13404–13405. doi:10.1021/ja076179.
- ↑ Reisman, S. E.; Doyle, A. G. (May 2008). "Enantioselective thiourea-catalyzed additions to oxocarbenium ions". J. Am. Chem. Soc. 130 (23): 7198–7199. PMC 2574628 . PMID 18479086. doi:10.1021/ja801514m.
- ↑ Zuend, S. J.; Jacobsen, E. N. (September 2009). "Mechanism of amido-thiourea catalyzed enantioselective imine hydrocyanation: transition state stabilization via multiple non-covalent interactions". J. Am. Chem. Soc. 131 (42): 15358–15374. PMC 2783581 . PMID 19778044. doi:10.1021/ja9058958.
- ↑ Uraguchi, D.; Terada, M. (April 2004). "Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation". J. Am. Chem. Soc. 126 (17): 5356–5357. PMID 15113196. doi:10.1021/ja0491533.
- ↑ Jansen, A. C. A.; Brussee, J. (May 1983). "A highly stereoselective synthesis of s(−)-[1,1'-binaphthalene]-2,2'-diol". Tet. Lett. 24 (31): 3261–3262. doi:10.1016/S0040-4039(00)88151-4.
- ↑ Lee, A.; Stewart, J. D.; Clardy, J.; Ganem, B. (April 1995). "New insight into the catalytic mechanism of chorismate mutases from structural studies". Chemistry & Biology. 2 (4): 195–203. doi:10.1016/1074-5521(95)90269-4.
- ↑ Knowles, R. R.; Lin, S.; Jacobsen, E. N. (April 2010). "Enantioselective thiourea-catalyzed cationic polycyclizations". J. Am. Chem. Soc. 132 (14): 5030–5032. PMC 2989498 . PMID 20369901. doi:10.1021/ja101256v.
- ↑ Wang, B.; Wu, F.; Wang, Y.; Liu, X.; Deng, L. (January 2007). "Control of diastereoselectivity in tandem asymmetric reactions generating nonadjacent stereocenters with bifunctional catalysis by Cinchona alkaloids". J. Am. Chem. Soc. 129 (4): 768–769. PMID 17243806. doi:10.1021/ja0670409.
- ↑ Wennemers, Helma (2011). "Asymmetric catalysis with peptides". Chem. Commun. 47: 12036–12041. doi:10.1039/C1CC15237H.
- ↑ Dalko, P. I. (2007). Enantioselective organocatalysis. Weinheim: Wiley-VCH. ISBN 978-3-527-31522-2.
- ↑ Schreiner, Peter R. (2003). "Metal-free organocatalysis through explicit hydrogen bonding interactions". Chem. Soc. Rev. 32 (5): 289–296. PMID 14518182. doi:10.1039/B107298F.
- ↑ Jacobsen, E. N. "Asymmetric catalysis with chiral H-bond donors" (PDF). Retrieved 18 December 2012.
- ↑ Jacobsen, E. N.; Dustin, J. M.; Zuend, S. J. (November 2008). "Catalytic asymmetric total synthesis of (+)-Yohimbine". Org. Lett. 10 (5): 745–748. PMID 18257582. doi:10.1021/ol702781q.
- ↑ Takemoto, Yoshiji; Miyabe, H. (July 2008). "Discovery and application of asymmetric reaction by multi-functional thioureas". Bull. Chem. Soc. Jap. 81 (7): 785–795. doi:10.1246/bcsj.81.785.
- ↑ Zuend, S. J.; Coughlin, M. P.; Lalonde, M. P.; Jacobsen, E. N. (October 2009). "Scaleable [sic] catalytic asymmetric Strecker syntheses of unnatural alpha-amino acids". Nature. 461 (7266): 968–970. PMC 2778849 . PMID 19829379. doi:10.1038/nature08484.