Homocystinuria
| Homocystinuria | |
|---|---|
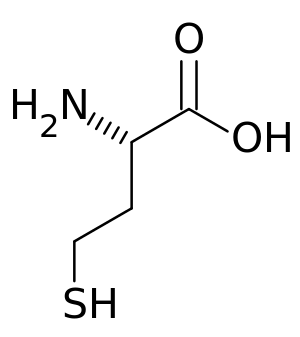 | |
| Homocysteine | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E72.1 |
| ICD-9-CM | 270.4 |
| OMIM | 236200 |
| DiseasesDB | 5991 |
| MedlinePlus | 001199 |
| eMedicine | derm/708 |
| Patient UK | Homocystinuria |
| MeSH | D006712 |
| GeneReviews | |
Classical homocystinuria, also known as cystathionine beta synthase deficiency or CBS deficiency,[1] is an inherited disorder of the metabolism of the amino acid methionine, often involving cystathionine beta synthase. It is an inherited autosomal recessive trait, which means a child needs to inherit a copy of the defective gene from both parents to be affected.
Signs and symptoms
This defect leads to a multi-systemic disorder of the connective tissue, muscles, central nervous system (CNS), and cardiovascular system. Homocystinuria represents a group of hereditary metabolic disorders characterized by an accumulation of the amino acid homocysteine in the serum and an increased excretion of homocysteine in the urine. Infants appear to be normal and early symptoms, if any are present, are vague.
Signs and symptoms of homocystinuria that may be seen include the following:
- A family history of homocystinuria[2]
- Flush across the cheeks
- Musculoskeletal
- Tall, thin build resembling Marfanoid habitus[1]
- Long limbs (dolichostenomelia)
- High-arched feet (pes cavus)
- Knock knees (genu valgum)
- Pectus excavatum and Pectus carinatum
- Intellectual disability
- Seizures
- Psychiatric disease
- Eye anomalies:
- Ectopia lentis – in contrast to Marfan syndrome which features upward ectopia lentis, downward dislocation is the typical finding of homocystinuria[3] or subluxation of lens
- Myopia (nearsightedness)
- Glaucoma
- Optic atrophy
- Retinal detachment[4]
- Cataracts
- Vascular disease
- Extensive atheroma formation at a young age which affects many arteries but not the coronary arteries
- Intravascular thrombosis
Cause
It is caused by the deficiency of the enzyme cystathionine beta synthase, and the deficiency of folic acid, vitamin B12 and pyridoxine (vitamin B6), or mutations of related enzymes.
Diagnosis
The term homocystinuria describes an increased excretion of the thiol amino acid homocysteine in urine (and incidentally, also an increased concentration in plasma). The source of this increase may be one of many metabolic factors, only one of which is CBS deficiency. Others include the re-methylation defects (cobalamin defects, methionine sythase deficiency, MTHFR) and vitamin deficiencies (cobalamin (vitamin B12) deficiency, folate (vitamin B9) deficiency, riboflavin deficiency (vitamin B2), pyridoxal phosphate deficiency (vitamin B6)). In light of this information, a combined approach to laboratory diagnosis is required to reach a differential diagnosis.
CBS deficiency may be diagnosed by routine metabolic biochemistry. In the first instance, plasma or urine amino acid analysis will frequently show an elevation of methionine and the presence of homocysteine. Many neonatal screening programs include methionine as a metabolite. The disorder may be distinguished from the re-methylation defects (e.g., MTHFR, methionine synthase deficiency and the cobalamin defects) in lieu of the elevated methionine concentration.[5] Additionally, organic acid analysis or quantitative determination of methylmalonic acid should help to exclude cobalamin (vitamin B12) defects and vitamin B12 deficiency giving a differential diagnosis.[6]
The laboratory analysis of homocysteine itself is complicated because most homocysteine (possibly above 85%) is bound to other thiol amino acids and proteins in the form of disulphides (e.g., cysteine in cystine-homocysteine, homocysteine in homocysteine-homocysteine) via disulfide bonds. Since as an equilibrium process the proportion of free homocystene is variable a true value of total homocysteine (free + bound) is useful for confirming diagnosis and particularly for monitoring of treatment efficacy. To this end it is prudent to perform total homocyst(e)ine analysis in which all disulphide bonds are subject to reduction prior to analysis, traditionally by HPLC after derivatisation with a fluorescent agent, thus giving a true reflection of the quantity of homocysteine in a plasma sample.[7]
Treatment
No specific cure has been discovered for homocystinuria; however, many people are treated using high doses of vitamin B6 (also known as pyridoxine).[8] Slightly less than 50% respond to this treatment and need to take supplemental vitamin B6 for the rest of their lives. Those who do not respond require a Low-sulfur diet (especially monitoring methionine), and most will need treatment with trimethylglycine. A normal dose of folic acid supplement and occasionally adding cysteine to the diet can be helpful, as glutathione is synthesized from cysteine (so adding cysteine can be important to reduce oxidative stress).
Betaine (N,N,N-trimethylglycine) is used to reduce concentrations of homocysteine by promoting the conversion of homocysteine back to methionine, i.e., increasing flux through the re-methylation pathway independent of folate derivatives (which is mainly active in the liver and in the kidneys).The re-formed methionine is then gradually removed by incorporation into body protein. The methionine that is not converted into protein is converted to S-adenosyl-methionine which goes on to form homocysteine again. Betaine is, therefore, only effective if the quantity of methionine to be removed is small. Hence treatment includes both betaine and a diet low in methionine. In classical homocystinuria (CBS, or cystathione beta synthase deficiency), the plasma methionine level usually increases above the normal range of 30 micromoles/L and the concentrations should be monitored as potentially toxic levels (more than 400 micromoles/L) may be reached.
Recommended diet
Low-protein food is recommended for this disorder, which requires food products low in particular types of amino acids (e.g., methionine).[9]
Prognosis
The life expectancy of patients with homocystinuria is reduced only if untreated. It is known that before the age of 30, almost one quarter of patients die as a result of thrombotic complications (e.g., heart attack).
Society and culture
One theory suggests that Akhenaten, a pharaoh of the eighteenth dynasty of Egypt, may have suffered from homocystinuria.[10]
See also
References
- 1 2 Online Mendelian Inheritance in Man (OMIM) 236200
- ↑ Maillot F, Kraus JP, Lee PJ (2008). "Environmental influences on familial discordance of phenotype in people with homocystinuria: a case report". J Med Case Reports. 2 (1): 113. PMC 2377250
 . PMID 18423051. doi:10.1186/1752-1947-2-113.
. PMID 18423051. doi:10.1186/1752-1947-2-113. - ↑ Peter Nicholas Robinson; Maurice Godfrey (2004). Marfan syndrome: a primer for clinicians and scientists. Springer. pp. 5–. ISBN 978-0-306-48238-0. Retrieved 12 April 2010.
- ↑ Goldman, Lee (2011). Goldman's Cecil Medicine (24th ed.). Philadelphia: Elsevier Saunders. p. 1362. ISBN 1437727883.
- ↑ (eds.), N. Blau ... (2003). Physician's guide to the laboratory diagnosis of metabolic diseases ; with 270 tables (2. ed.). Berlin [u.a.]: Springer. ISBN 354042542X.
- ↑ Refsum, Helga; A. David Smith; Per M. Ueland; Ebba Nexo; Robert Clarke; Joseph McPartlin; Carole Johnston; Frode Engbaek; Jørn Schneede; Catherine McPartlin; John M. Scott (2004). "Facts and Recommendations about Total Homocysteine Determinations: An Expert Opinion". Clinical Chemistry. 50 (1): 3–32. PMID 14709635. doi:10.1373/clinchem.2003.021634.
- ↑ Carducci, Claudia; M. Birarelli; M. Nola; I. Antonozzi (1999). "Automated high-performance liquid chromatographic method for the determination of homocysteine in plasma samples". Journal of Chromatography A. 846 (1–2): 93–100. PMID 10420601. doi:10.1016/S0021-9673(98)01091-7.
- ↑ Bakker, R. C.; Brandjes, D. P. (June 1997). "Hyperhomocysteinaemia and associated disease". Pharmacy world & science : PWS. 19 (3): 126–132. PMID 9259028. doi:10.1023/A:1008634632501.
- ↑ YAP, SUFIN; NAUGHTEN, EILEEN R.; WILCKEN, BRIDGET; WILCKEN, DAVID E.L.; BOERS, GODFRIED H.J. (2000-01-01). "Vascular Complications of Severe Hyperhomocysteinemia in Patients with Homocystinuria Due to Cystathionine β-Synthase Deficiency: Effects of Homocysteine-Lowering Therapy". Seminars in Thrombosis and Hemostasis. 26 (03): 335–340. ISSN 0094-6176. doi:10.1055/s-2000-8100.
- ↑ Cavka M, Kelava T (Mar 2010). "Homocystinuria, a possible solution of the Akhenaten's mystery". Coll Antropol. 34: 255–58. PMID 20402329.
External links
- Homocystinuria Support
- GeneReview/NIH/UW entry on Homocystinuria Caused by Cystathionine Beta-Synthase Deficiency
- Paper and discussion on Homocystinuria due to Cystathionine Beta Synthase deficiency