High-performance liquid chromatography
 An HPLC. From left to right: A pumping device generating a gradient of two different solvents- a steel-enforced column and a detector for measuring the absorbance. | |
| Acronym | HPLC |
|---|---|
| Classification | Chromatography |
| Analytes |
organic molecules biomolecules ions polymers |
| Other techniques | |
| Related |
Chromatography Aqueous normal-phase chromatography Hydrophilic Interaction Chromatography Ion exchange chromatography Size exclusion chromatography Micellar liquid chromatography |
| Hyphenated | Liquid chromatography-mass spectrometry |

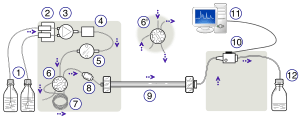
High-performance liquid chromatography (HPLC; formerly referred to as high-pressure liquid chromatography), is a technique in analytical chemistry used to separate, identify, and quantify each component in a mixture. It relies on pumps to pass a pressurized liquid solvent containing the sample mixture through a column filled with a solid adsorbent material. Each component in the sample interacts slightly differently with the adsorbent material, causing different flow rates for the different components and leading to the separation of the components as they flow out the column.
HPLC has been used for manufacturing (e.g. during the production process of pharmaceutical and biological products), legal (e.g. detecting performance enhancement drugs in urine), research (e.g. separating the components of a complex biological sample, or of similar synthetic chemicals from each other), and medical (e.g. detecting vitamin D levels in blood serum) purposes.[1]
Chromatography can be described as a mass transfer process involving adsorption. HPLC relies on pumps to pass a pressurized liquid and a sample mixture through a column filled with adsorbent, leading to the separation of the sample components. The active component of the column, the adsorbent, is typically a granular material made of solid particles (e.g. silica, polymers, etc.), 2–50 μm in size. The components of the sample mixture are separated from each other due to their different degrees of interaction with the adsorbent particles. The pressurized liquid is typically a mixture of solvents (e.g. water, acetonitrile and/or methanol) and is referred to as a "mobile phase". Its composition and temperature play a major role in the separation process by influencing the interactions taking place between sample components and adsorbent. These interactions are physical in nature, such as hydrophobic (dispersive), dipole–dipole and ionic, most often a combination.
HPLC is distinguished from traditional ("low pressure") liquid chromatography because operational pressures are significantly higher (50–350 bar), while ordinary liquid chromatography typically relies on the force of gravity to pass the mobile phase through the column. Due to the small sample amount separated in analytical HPLC, typical column dimensions are 2.1–4.6 mm diameter, and 30–250 mm length. Also HPLC columns are made with smaller sorbent particles (2–50 μm in average particle size). This gives HPLC superior resolving power (the ability to distinguish between compounds) when separating mixtures, which makes it a popular chromatographic technique.
The schematic of an HPLC instrument typically includes a degasser, sampler, pumps, and a detector. The sampler brings the sample mixture into the mobile phase stream which carries it into the column. The pumps deliver the desired flow and composition of the mobile phase through the column. The detector generates a signal proportional to the amount of sample component emerging from the column, hence allowing for quantitative analysis of the sample components. A digital microprocessor and user software control the HPLC instrument and provide data analysis. Some models of mechanical pumps in a HPLC instrument can mix multiple solvents together in ratios changing in time, generating a composition gradient in the mobile phase. Various detectors are in common use, such as UV/Vis, photodiode array (PDA) or based on mass spectrometry. Most HPLC instruments also have a column oven that allows for adjusting the temperature at which the separation is performed.
Operation
The sample mixture to be separated and analyzed is introduced, in a discrete small volume (typically microliters), into the stream of mobile phase percolating through the column. The components of the sample move through the column at different velocities, which are a function of specific physical interactions with the adsorbent (also called stationary phase). The velocity of each component depends on its chemical nature, on the nature of the stationary phase (column) and on the composition of the mobile phase. The time at which a specific analyte elutes (emerges from the column) is called its retention time. The retention time measured under particular conditions is an identifying characteristic of a given analyte.
Many different types of columns are available, filled with adsorbents varying in particle size, and in the nature of their surface ("surface chemistry"). The use of smaller particle size packing materials requires the use of higher operational pressure ("backpressure") and typically improves chromatographic resolution (i.e. the degree of separation between consecutive analytes emerging from the column). Sorbent particles may be hydrophobic or polar in nature.
Common mobile phases used include any miscible combination of water with various organic solvents (the most common are acetonitrile and methanol). Some HPLC techniques use water-free mobile phases (see Normal-phase chromatography below). The aqueous component of the mobile phase may contain acids (such as formic, phosphoric or trifluoroacetic acid) or salts to assist in the separation of the sample components. The composition of the mobile phase may be kept constant ("isocratic elution mode") or varied ("gradient elution mode") during the chromatographic analysis. Isocratic elution is typically effective in the separation of sample components that are very different in their affinity for the stationary phase. In gradient elution the composition of the mobile phase is varied typically from low to high eluting strength. The eluting strength of the mobile phase is reflected by analyte retention times with high eluting strength producing fast elution (=short retention times). A typical gradient profile in reversed phase chromatography might start at 5% acetonitrile (in water or aqueous buffer) and progress linearly to 95% acetonitrile over 5–25 minutes. Periods of constant mobile phase composition may be part of any gradient profile. For example, the mobile phase composition may be kept constant at 5% acetonitrile for 1–3 min, followed by a linear change up to 95% acetonitrile.

The chosen composition of the mobile phase (also called eluent) depends on the intensity of interactions between various sample components ("analytes") and stationary phase (e.g. hydrophobic interactions in reversed-phase HPLC). Depending on their affinity for the stationary and mobile phases analytes partition between the two during the separation process taking place in the column. This partitioning process is similar to that which occurs during a liquid–liquid extraction but is continuous, not step-wise. In this example, using a water/acetonitrile gradient, more hydrophobic components will elute (come off the column) late, once the mobile phase gets more concentrated in acetonitrile (i.e. in a mobile phase of higher eluting strength).
The choice of mobile phase components, additives (such as salts or acids) and gradient conditions depends on the nature of the column and sample components. Often a series of trial runs is performed with the sample in order to find the HPLC method which gives adequate separation.
History and development
Prior to HPLC scientists used standard liquid chromatographic techniques. Liquid chromatographic systems were largely inefficient due to the flow rate of solvents being dependent on gravity. Separations took many hours, and sometimes days to complete. Gas chromatography (GC) at the time was more powerful than liquid chromatography (LC), however, it was believed that gas phase separation and analysis of very polar high molecular weight biopolymers was impossible.[3] GC was ineffective for many biochemists because of the thermal instability of the solutes.[4] As a result, alternative methods were hypothesized which would soon result in the development of HPLC.
Following on the seminal work of Martin and Synge in 1941, it was predicted by Cal Giddings, Josef Huber, and others in the 1960s that LC could be operated in the high-efficiency mode by reducing the packing-particle diameter substantially below the typical LC (and GC) level of 150 μm and using pressure to increase the mobile phase velocity.[3] These predictions underwent extensive experimentation and refinement throughout the 60s into the 70s. Early developmental research began to improve LC particles, and the invention of Zipax, a superficially porous particle, was promising for HPLC technology.[5]
The 1970s brought about many developments in hardware and instrumentation. Researchers began using pumps and injectors to make a rudimentary design of an HPLC system.[6] Gas amplifier pumps were ideal because they operated at constant pressure and did not require leak free seals or check valves for steady flow and good quantitation.[4] Hardware milestones were made at Dupont IPD (Industrial Polymers Division) such as a low-dwell-volume gradient device being utilized as well as replacing the septum injector with a loop injection valve.[4]
While instrumentational developments were important, the history of HPLC is primarily about the history and evolution of particle technology.[4] After the introduction of porous layer particles, there has been a steady trend to reduced particle size to improve efficiency.[4] However, by decreasing particle size, new problems arose. The practical disadvantages stem from the excessive pressure drop needed to force mobile fluid through the column and the difficulty of preparing a uniform packing of extremely fine materials.[7] Every time particle size is reduced significantly, another round of instrument development usually must occur to handle the pressure.[4]
Types
Partition chromatography

Partition chromatography was one of the first kinds of chromatography that chemists developed.[8] The partition coefficient principle has been applied in paper chromatography, thin layer chromatography, gas phase and liquid–liquid separation applications. The 1952 Nobel Prize in chemistry was earned by Archer John Porter Martin and Richard Laurence Millington Synge for their development of the technique, which was used for their separation of amino acids.[9] Partition chromatography uses a retained solvent, on the surface or within the grains or fibers of an "inert" solid supporting matrix as with paper chromatography; or takes advantage of some coulombic and/or hydrogen donor interaction with the stationary phase. Analyte molecules partition between a liquid stationary phase and the eluent. Just as in Hydrophilic Interaction Chromatography (HILIC; a sub-technique within HPLC), this method separates analytes based on differences in their polarity. HILIC most often uses a bonded polar stationary phase and a mobile phase made primarily of acetonitrile with water as the strong component. Partition HPLC has been used historically on unbonded silica or alumina supports. Each works effectively for separating analytes by relative polar differences. HILIC bonded phases have the advantage of separating acidic, basic and neutral solutes in a single chromatographic run.[10]
The polar analytes diffuse into a stationary water layer associated with the polar stationary phase and are thus retained. The stronger the interactions between the polar analyte and the polar stationary phase (relative to the mobile phase) the longer the elution time. The interaction strength depends on the functional groups part of the analyte molecular structure, with more polarized groups (e.g. hydroxyl-) and groups capable of hydrogen bonding inducing more retention. Coulombic (electrostatic) interactions can also increase retention. Use of more polar solvents in the mobile phase will decrease the retention time of the analytes, whereas more hydrophobic solvents tend to increase retention times.
Normal–phase chromatography
Normal–phase chromatography was one of the first kinds of HPLC that chemists developed. Also known as normal-phase HPLC (NP-HPLC) this method separates analytes based on their affinity for a polar stationary surface such as silica, hence it is based on analyte ability to engage in polar interactions (such as hydrogen-bonding or dipole-dipole type of interactions) with the sorbent surface. NP-HPLC uses a non-polar, non-aqueous mobile phase (e.g. Chloroform), and works effectively for separating analytes readily soluble in non-polar solvents. The analyte associates with and is retained by the polar stationary phase. Adsorption strengths increase with increased analyte polarity. The interaction strength depends not only on the functional groups present in the structure of the analyte molecule, but also on steric factors. The effect of steric hindrance on interaction strength allows this method to resolve (separate) structural isomers.
The use of more polar solvents in the mobile phase will decrease the retention time of analytes, whereas more hydrophobic solvents tend to induce slower elution (increased retention times). Very polar solvents such as traces of water in the mobile phase tend to adsorb to the solid surface of the stationary phase forming a stationary bound (water) layer which is considered to play an active role in retention. This behavior is somewhat peculiar to normal phase chromatography because it is governed almost exclusively by an adsorptive mechanism (i.e. analytes interact with a solid surface rather than with the solvated layer of a ligand attached to the sorbent surface; see also reversed-phase HPLC below). Adsorption chromatography is still widely used for structural isomer separations in both column and thin-layer chromatography formats on activated (dried) silica or alumina supports.
Partition- and NP-HPLC fell out of favor in the 1970s with the development of reversed-phase HPLC because of poor reproducibility of retention times due to the presence of a water or protic organic solvent layer on the surface of the silica or alumina chromatographic media. This layer changes with any changes in the composition of the mobile phase (e.g. moisture level) causing drifting retention times.
Recently, partition chromatography has become popular again with the development of Hilic bonded phases which demonstrate improved reproducibility, and due to a better understanding of the range of usefulness of the technique.
Displacement chromatography
The basic principle of displacement chromatography is: A molecule with a high affinity for the chromatography matrix (the displacer) will compete effectively for binding sites, and thus displace all molecules with lesser affinities.[11] There are distinct differences between displacement and elution chromatography. In elution mode, substances typically emerge from a column in narrow, Gaussian peaks. Wide separation of peaks, preferably to baseline, is desired in order to achieve maximum purification. The speed at which any component of a mixture travels down the column in elution mode depends on many factors. But for two substances to travel at different speeds, and thereby be resolved, there must be substantial differences in some interaction between the biomolecules and the chromatography matrix. Operating parameters are adjusted to maximize the effect of this difference. In many cases, baseline separation of the peaks can be achieved only with gradient elution and low column loadings. Thus, two drawbacks to elution mode chromatography, especially at the preparative scale, are operational complexity, due to gradient solvent pumping, and low throughput, due to low column loadings. Displacement chromatography has advantages over elution chromatography in that components are resolved into consecutive zones of pure substances rather than “peaks”. Because the process takes advantage of the nonlinearity of the isotherms, a larger column feed can be separated on a given column with the purified components recovered at significantly higher concentration.
Reversed-phase chromatography (RPC)

Reversed phase HPLC (RP-HPLC) has a non-polar stationary phase and an aqueous, moderately polar mobile phase. One common stationary phase is a silica which has been surface-modified with RMe2SiCl, where R is a straight chain alkyl group such as C18H37 or C8H17. With such stationary phases, retention time is longer for molecules which are less polar, while polar molecules elute more readily (early in the analysis). An investigator can increase retention times by adding more water to the mobile phase; thereby making the affinity of the hydrophobic analyte for the hydrophobic stationary phase stronger relative to the now more hydrophilic mobile phase. Similarly, an investigator can decrease retention time by adding more organic solvent to the eluent. RP-HPLC is so commonly used that it is often incorrectly referred to as "HPLC" without further specification. The pharmaceutical industry regularly employs RP-HPLC to qualify drugs before their release.
RP-HPLC operates on the principle of hydrophobic interactions, which originates from the high symmetry in the dipolar water structure and plays the most important role in all processes in life science. RP-HPLC allows the measurement of these interactive forces. The binding of the analyte to the stationary phase is proportional to the contact surface area around the non-polar segment of the analyte molecule upon association with the ligand on the stationary phase. This solvophobic effect is dominated by the force of water for "cavity-reduction" around the analyte and the C18-chain versus the complex of both. The energy released in this process is proportional to the surface tension of the eluent (water: 7.3×10−6 J/cm², methanol: 2.2×10−6 J/cm²) and to the hydrophobic surface of the analyte and the ligand respectively. The retention can be decreased by adding a less polar solvent (methanol, acetonitrile) into the mobile phase to reduce the surface tension of water. Gradient elution uses this effect by automatically reducing the polarity and the surface tension of the aqueous mobile phase during the course of the analysis.
Structural properties of the analyte molecule play an important role in its retention characteristics. In general, an analyte with a larger hydrophobic surface area (C–H, C–C, and generally non-polar atomic bonds, such as S-S and others) is retained longer because it is non-interacting with the water structure. On the other hand, analytes with higher polar surface area (conferred by the presence of polar groups, such as -OH, -NH2, COO− or -NH3+ in their structure) are less retained as they are better integrated into water. Such interactions are subject to steric effects in that very large molecules may have only restricted access to the pores of the stationary phase, where the interactions with surface ligands (alkyl chains) take place. Such surface hindrance typically results in less retention.
Retention time increases with hydrophobic (non-polar) surface area. Branched chain compounds elute more rapidly than their corresponding linear isomers because the overall surface area is decreased. Similarly organic compounds with single C–C bonds elute later than those with a C=C or C–C triple bond, as the double or triple bond is shorter than a single C–C bond.
Aside from mobile phase surface tension (organizational strength in eluent structure), other mobile phase modifiers can affect analyte retention. For example, the addition of inorganic salts causes a moderate linear increase in the surface tension of aqueous solutions (ca. 1.5×10−7 J/cm² per Mol for NaCl, 2.5×10−7 J/cm² per Mol for (NH4)2SO4), and because the entropy of the analyte-solvent interface is controlled by surface tension, the addition of salts tend to increase the retention time. This technique is used for mild separation and recovery of proteins and protection of their biological activity in protein analysis (hydrophobic interaction chromatography, HIC).
Another important factor is the mobile phase pH since it can change the hydrophobic character of the analyte. For this reason most methods use a buffering agent, such as sodium phosphate, to control the pH. Buffers serve multiple purposes: control of pH, neutralize the charge on the silica surface of the stationary phase and act as ion pairing agents to neutralize analyte charge. Ammonium formate is commonly added in mass spectrometry to improve detection of certain analytes by the formation of analyte-ammonium adducts. A volatile organic acid such as acetic acid, or most commonly formic acid, is often added to the mobile phase if mass spectrometry is used to analyze the column effluent. Trifluoroacetic acid is used infrequently in mass spectrometry applications due to its persistence in the detector and solvent delivery system, but can be effective in improving retention of analytes such as carboxylic acids in applications utilizing other detectors, as it is a fairly strong organic acid. The effects of acids and buffers vary by application but generally improve chromatographic resolution.
Reversed phase columns are quite difficult to damage compared with normal silica columns; however, many reversed phase columns consist of alkyl derivatized silica particles and should never be used with aqueous bases as these will destroy the underlying silica particle. They can be used with aqueous acid, but the column should not be exposed to the acid for too long, as it can corrode the metal parts of the HPLC equipment. RP-HPLC columns should be flushed with clean solvent after use to remove residual acids or buffers, and stored in an appropriate composition of solvent. The metal content of HPLC columns must be kept low if the best possible ability to separate substances is to be retained. A good test for the metal content of a column is to inject a sample which is a mixture of 2,2'- and 4,4'- bipyridine. Because the 2,2'-bipy can chelate the metal, the shape of the peak for the 2,2'-bipy will be distorted (tailed) when metal ions are present on the surface of the silica...
Size-exclusion chromatography
Size-exclusion chromatography (SEC), also known as gel permeation chromatography or gel filtration chromatography, separates particles on the basis of molecular size (actually by a particle's Stokes radius). It is generally a low resolution chromatography and thus it is often reserved for the final, "polishing" step of the purification. It is also useful for determining the tertiary structure and quaternary structure of purified proteins. SEC is used primarily for the analysis of large molecules such as proteins or polymers. SEC works by trapping these smaller molecules in the pores of a particle. The larger molecules simply pass by the pores as they are too large to enter the pores. Larger molecules therefore flow through the column quicker than smaller molecules, that is, the smaller the molecule, the longer the retention time.
This technique is widely used for the molecular weight determination of polysaccharides. SEC is the official technique (suggested by European pharmacopeia) for the molecular weight comparison of different commercially available low-molecular weight heparins.
Ion-exchange chromatography
In ion-exchange chromatography (IC), retention is based on the attraction between solute ions and charged sites bound to the stationary phase. Solute ions of the same charge as the charged sites on the column are excluded from binding, while solute ions of the opposite charge of the charged sites of the column are retained on the column. Solute ions that are retained on the column can be eluted from the column by changing the solvent conditions (e.g. increasing the ion effect of the solvent system by increasing the salt concentration of the solution, increasing the column temperature, changing the pH of the solvent, etc...).
Types of ion exchangers include:
- Polystyrene resins – These allow cross linkage which increases the stability of the chain. Higher cross linkage reduces swerving, which increases the equilibration time and ultimately improves selectivity.
- Cellulose and dextran ion exchangers (gels) – These possess larger pore sizes and low charge densities making them suitable for protein separation.
- Controlled-pore glass or porous silica
In general, ion exchangers favor the binding of ions of higher charge and smaller radius.
An increase in counter ion (with respect to the functional groups in resins) concentration reduces the retention time. A decrease in pH reduces the retention time in cation exchange while an increase in pH reduces the retention time in anion exchange. By lowering the pH of the solvent in a cation exchange column, for instance, more hydrogen ions are available to compete for positions on the anionic stationary phase, thereby eluting weakly bound cations.
This form of chromatography is widely used in the following applications: water purification, preconcentration of trace components, ligand-exchange chromatography, ion-exchange chromatography of proteins, high-pH anion-exchange chromatography of carbohydrates and oligosaccharides, and others.
Bioaffinity chromatography
This chromatographic process relies on the property of biologically active substances to form stable, specific, and reversible complexes. The formation of these complexes involves the participation of common molecular forces such as the Van der Waals interaction, electrostatic interaction, dipole-dipole interaction, hydrophobic interaction, and the hydrogen bond. An efficient, biospecific bond is formed by a simultaneous and concerted action of several of these forces in the complementary binding sites.
Aqueous normal-phase chromatography
Aqueous normal-phase chromatography (ANP) is a chromatographic technique which encompasses the mobile phase region between reversed-phase chromatography (RP) and organic normal phase chromatography (ONP). This technique is used to achieve unique selectivity for hydrophilic compounds, showing normal phase elution using reversed-phase solvents.
Isocratic and gradient elution

A separation in which the mobile phase composition remains constant throughout the procedure is termed isocratic (meaning constant composition). The word was coined by Csaba Horvath who was one of the pioneers of HPLC.,
The mobile phase composition does not have to remain constant. A separation in which the mobile phase composition is changed during the separation process is described as a gradient elution.[12] One example is a gradient starting at 10% methanol and ending at 90% methanol after 20 minutes. The two components of the mobile phase are typically termed "A" and "B"; A is the "weak" solvent which allows the solute to elute only slowly, while B is the "strong" solvent which rapidly elutes the solutes from the column. In reversed-phase chromatography, solvent A is often water or an aqueous buffer, while B is an organic solvent miscible with water, such as acetonitrile, methanol, THF, or isopropanol.
In isocratic elution, peak width increases with retention time linearly according to the equation for N, the number of theoretical plates. This leads to the disadvantage that late-eluting peaks get very flat and broad. Their shape and width may keep them from being recognized as peaks.
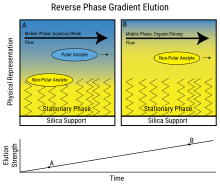
Gradient elution decreases the retention of the later-eluting components so that they elute faster, giving narrower (and taller) peaks for most components. This also improves the peak shape for tailed peaks, as the increasing concentration of the organic eluent pushes the tailing part of a peak forward. This also increases the peak height (the peak looks "sharper"), which is important in trace analysis. The gradient program may include sudden "step" increases in the percentage of the organic component, or different slopes at different times – all according to the desire for optimum separation in minimum time.
In isocratic elution, the selectivity does not change if the column dimensions (length and inner diameter) change – that is, the peaks elute in the same order. In gradient elution, the elution order may change as the dimensions or flow rate change.
The driving force in reversed phase chromatography originates in the high order of the water structure. The role of the organic component of the mobile phase is to reduce this high order and thus reduce the retarding strength of the aqueous component.
Parameters
Theoretical
HPLC separations have theoretical parameters and equations to describe the separation of components into signal peaks when detected by instrumentation such as by a UV detector or a mass spectrometer. The parameters are largely derived from two sets of chromatagraphic theory: plate theory (as part of Partition chromatography), and the rate theory of chromatography / Van Deemter equation. Of course, they can be put in practice through analysis of HPLC chromatograms, although rate theory is considered the more accurate theory.
They are analogous to the calculation of retention factor for a paper chromatography separation, but describes how well HPLC separates a mixture into two or more components that are detected as peaks (bands) on a chromatogram. The HPLC parameters are the: efficiency factor(N), the retention factor (kappa prime), and the separation factor (alpha). Together the factors are variables in a resolution equation, which describes how well two components' peaks separated or overlapped each other. These parameters are mostly only used for describing HPLC reversed phase and HPLC normal phase separations, since those separations tend to be more subtle than other HPLC modes (e.g. ion exchange and size exclusion).
- Void volume is the amount of space in a column that is occupied by solvent. It is the space within the column that is outside of the column's internal packing material. Void volume is measured on a chromatogram as the first component peak detected, which is usually the solvent that was present in the sample mixture; ideally the sample solvent flows through the column without interacting with the column, but is still detectable as distinct from the HPLC solvent. The void volume is used as a correction factor.
- Efficiency factor (N) practically measures how sharp component peaks on the chromatogram are, as ratio of the component peak's area ("retention time") relative to the width of the peaks at their widest point (at the baseline). Peaks that are tall, sharp, and relatively narrow indicate that separation method efficiently removed a component from a mixture; high efficiency. Efficiency is very dependent upon the HPLC column and the HPLC method used. Efficiency factor is synonymous with plate number, and the 'number of theoretical plates'.
- Retention factor (kappa prime) measures how long a component of the mixture stuck to the column, measured by the area under the curve of its peak in a chromatogram (since HPLC chromatograms are a function of time). Each chromatogram peak will have its own retention factor (e.g. kappa1 for the retention factor of the first peak). This factor may be corrected for by the void volume of the column.
- Separation factor (alpha) is a relative comparison on how well two neighboring components of the mixture were separated (i.e. two neighboring bands on a chromatogram). This factor is defined in terms of a ratio of the retention factors of a pair of neighboring chromatogram peaks, and may also be corrected for by the void volume of the column. The greater the separation factor value is over 1.0, the better the separation, until about 2.0 beyond which an HPLC method is probably not needed for separation.
Resolution equations relate the three factors such that high efficiency and separation factors improve the resolution of component peaks in a HPLC separation.
Internal diameter
.jpg)
The internal diameter (ID) of an HPLC column is an important parameter that influences the detection sensitivity and separation selectivity in gradient elution. It also determines the quantity of analyte that can be loaded onto the column. Larger columns are usually seen in industrial applications, such as the purification of a drug product for later use. Low-ID columns have improved sensitivity and lower solvent consumption at the expense of loading capacity.
- Larger ID columns (over 10 mm) are used to purify usable amounts of material because of their large loading capacity.
- Analytical scale columns (4.6 mm) have been the most common type of columns, though smaller columns are rapidly gaining in popularity. They are used in traditional quantitative analysis of samples and often use a UV-Vis absorbance detector.
- Narrow-bore columns (1–2 mm) are used for applications when more sensitivity is desired either with special UV-vis detectors, fluorescence detection or with other detection methods like liquid chromatography-mass spectrometry
- Capillary columns (under 0.3 mm) are used almost exclusively with alternative detection means such as mass spectrometry. They are usually made from fused silica capillaries, rather than the stainless steel tubing that larger columns employ.
Particle size
Most traditional HPLC is performed with the stationary phase attached to the outside of small spherical silica particles (very small beads). These particles come in a variety of sizes with 5 µm beads being the most common. Smaller particles generally provide more surface area and better separations, but the pressure required for optimum linear velocity increases by the inverse of the particle diameter squared.[13][14][15]
This means that changing to particles that are half as big, keeping the size of the column the same, will double the performance, but increase the required pressure by a factor of four. Larger particles are used in preparative HPLC (column diameters 5 cm up to >30 cm) and for non-HPLC applications such as solid-phase extraction.
Pore size
Many stationary phases are porous to provide greater surface area. Small pores provide greater surface area while larger pore size has better kinetics, especially for larger analytes. For example, a protein which is only slightly smaller than a pore might enter the pore but does not easily leave once inside.
Pump pressure
Pumps vary in pressure capacity, but their performance is measured on their ability to yield a consistent and reproducible volumetric flow rate. Pressure may reach as high as 60 MPa (6000 lbf/in2), or about 600 atmospheres. Modern HPLC systems have been improved to work at much higher pressures, and therefore are able to use much smaller particle sizes in the columns (<2 μm). These "Ultra High Performance Liquid Chromatography" systems or UHPLCs can work at up to 120 MPa (17,405 lbf/in2), or about 1200 atmospheres.[16] The term "UPLC" is a trademark of the Waters Corporation, but is sometimes used to refer to the more general technique of UHPLC.
Detectors
HPLC most commonly uses a UV-Vis absorbance detector, however, a wide range of other chromatography detectors can be used. A universal detector that complements UV-Vis absorbance detection is the Charged aerosol detector (CAD). A kind of commonly utilized detector includes refractive index detectors, which provide readings by measuring the changes in the refractive index of the effluent as it moves through the flow cell. In certain cases, it is possible to use multiple detectors, for example LCMS normally combines UV-Vis with a mass spectrometer.
Autosamplers
Large numbers of samples can be automatically injected onto an HPLC system, by the use of HPLC autosamplers. In addition, HPLC autosamplers have an injection volume and technique which is exactly the same for each injection, consequently they provide a high degree of injection volume precision.
Applications
Manufacturing
As briefly mentioned, HPLC has many applications in both laboratory and clinical science. It is a common technique used in pharmaceutical development, as it is a dependable way to obtain and ensure product purity.[17] While HPLC can produce extremely high quality (pure) products, it is not always the primary method used in the production of bulk drug materials.[18] According to the European pharmacopoeia, HPLC is used in only 15.5% of syntheses.[19] However, it plays a role in 44% of syntheses in the United States pharmacopoeia.[20] This could possibly be due to differences in monetary and time constraints, as HPLC on a large scale can be an expensive technique. An increase in specificity, precision, and accuracy that occurs with HPLC unfortunately corresponds to an increase in cost.
Legal
This technique is also used for detection of illicit drugs in urine. The most common method of drug detection is an immunoassay.[21] This method is much more convenient. However, convenience comes at the cost of specificity and coverage of a wide range of drugs. As HPLC is a method of determining (and possibly increasing) purity, using HPLC alone in evaluating concentrations of drugs is somewhat insufficient. With this, HPLC in this context is often performed in conjunction with mass spectrometry.[22] Using liquid chromatography instead of gas chromatography in conjunction with MS circumvents the necessity for derivitizing with acetylating or alkylation agents, which can be a burdensome extra step.[23] This technique has been used to detect a variety of agents like doping agents, drug metabolites, glucuronide conjugates, amphetamines, opioids, cocaine, BZDs, ketamine, LSD, cannabis, and pesticides[24][25] Performing HPLC in conjunction with Mass spectrometry reduces the absolute need for standardizing HPLC experimental runs.
Research
Similar assays can be performed for research purposes, detecting concentrations of potential clinical candidates like anti-fungal and asthma drugs.[26] This technique is obviously useful in observing multiple species in collected samples, as well, but requires the use of standard solutions when information about species identity is sought out. It is used as a method to confirm results of synthesis reactions, as purity is essential in this type of research. However, mass spectrometry is still the more reliable way to identify species.
Medical
Medical use of HPLC can include drug analysis, but falls more closely under the category of nutrient analysis. While urine is the most common medium for analyzing drug concentrations, blood serum is the sample collected for most medical analyses with HPLC.[27] Other methods of detection of molecules that are useful for clinical studies have been tested against HPLC, namely immunoassays. In one example of this, competitive protein binding assays (CPBA) and HPLC were compared for sensitivity in detection of vitamin D. Useful for diagnosing vitamin D deficiencies in children, it was found that sensitivity and specificity of this CPBA reached only 40% and 60%, respectively, of the capacity of HPLC.[28] While an expensive tool, the accuracy of HPLC is nearly unparalleled.
See also
- History of chromatography
- Csaba Horváth
- Ion chromatography
- Column chromatography
- Capillary electrochromatography
- Micellar liquid chromatography
References
- ↑ Gerber, F.; Krummen, M.; Potgeter, H.; Roth, A.; Siffrin, C.; Spoendlin, C. (2004). "Practical aspects of fast reversed-phase high-performance liquid chromatography using 3μm particle packed columns and monolithic columns in pharmaceutical development and production working under current good manufacturing practice". Journal of Chromatography A. 1036 (2): 127–133. PMID 15146913. doi:10.1016/j.chroma.2004.02.056.
- ↑ Morgan, David J. "Fraction collector (post on Flickr)". Flickr. Retrieved 28 October 2015.
- 1 2 Karger, Barry L. (1997). "HPLC: Early and Recent Perspectives". Journal of Chemical Education. 74: 45. doi:10.1021/ed074p45.
- 1 2 3 4 5 6 Henry, Richard A. (1 February 2009) "The Early Days of HPLC at Dupont". Chromatography Online. Avanstar Communications Inc.
- ↑ Iler, R.K. (1979) The Chemistry of Silica. John Wiley & Sons. New York.
- ↑ Karger, B. L.; Berry, L. V. (1971). "Rapid liquid-chromatographic separation of steroids on columns heavily loaded with stationary phase". Clin. Chem. 17 (8): 757–64. PMID 4254537.
- ↑ Giddings, J. Calvin (1965) Dynamics of Chromatography, Part I. Principles and Theory. Marcel Dekker, Inc., New York. p. 281.
- ↑ Ettre, C. (2001). "Milestones in Chromatography: The Birth of Partition Chromatography" (PDF). LCGC. 19 (5): 506–512. Retrieved 2016-02-26.
- ↑ Martin, A J P; Synge, R L M (1941). "Separation of the higher monoamino-acids by counter-current liquid-liquid extraction: the amino-acid composition of wool". Biochemical Journal. 35 (1–2): 91–121. PMC 1265473
 . PMID 16747393. doi:10.1042/bj0350091.
. PMID 16747393. doi:10.1042/bj0350091. - ↑ Lindsay, S.; Kealey, D. (1987). High performance liquid chromatography. Wiley. from review Hung, L. B.; Parcher, J. F.; Shores, J. C.; Ward, E. H. (1988). "Theoretical and experimental foundation for surface-coverage programming in gas–solid chromatography with an adsorbable carrier gas". J. Am. Chem. Soc. 110 (11): 1090–1096. doi:10.1021/ac00162a003.
- ↑ Displacement Chromatography. Sacheminc.com. Retrieved 2011-06-07. Archived September 15, 2008, at the Wayback Machine.
- ↑ Snyder, Lloyd R.; Dolan, John W. (2006). High-Performance Gradient Elution: The Practical Application of the Linear-Solvent-Strength Model. Wiley Interscience. ISBN 0470055510.
- ↑ Majors, Ronald E.. (2010-09-07) Fast and Ultrafast HPLC on sub-2 μm Porous Particles — Where Do We Go From Here? – LC-GC Europe. Lcgceurope.com. Retrieved 2011-06-07.
- ↑ Xiang, Y.; Liu Y.; Lee M.L. (2006). "Ultrahigh pressure liquid chromatography using elevated temperature". Journal of Chromatography A. 1104 (1–2): 198–202. PMID 16376355. doi:10.1016/j.chroma.2005.11.118.
- ↑ Horváth, Cs.; Preiss B.A.; Lipsky S.R. (1967). "Fast liquid chromatography. Investigation of operating parameters and the separation of nucleotides on pellicular ion exchangers". Analytical Chemistry. 39 (12): 1422–1428. PMID 6073805. doi:10.1021/ac60256a003.
- ↑ 1290 Infinity Quaternary Pump. Agilent
- ↑ Gerber, Frederic (May 2004). "Practical aspects of fast reversed-phase high-performance liquid chromatography using 3 μm particle packed columns and monolithic columns in pharmaceutical development and production working under current good manufacturing practice". Journal of Chromatography. 1036 (2): 127–33. PMID 15146913. doi:10.1016/j.chroma.2004.02.056.
- ↑ Siddiqui, Masoom Raza; AlOthman, Zeid A.; Rahman, Nafisur (2013). "Analytical techniques in pharmaceutical analysis: A review". Arabian Journal of Chemistry. 10: S1409. doi:10.1016/j.arabjc.2013.04.016.
- ↑ The European Pharmacopoeia, 2002. fourth ed., Council of Europe, Strasbourg.
- ↑ United States Pharmacopoeia, 2004. 27th ed. The USP Convention Inc., Rockville, MD.
- ↑ Pesce, Amadeo; Rosenthal, Murray; West, Robert; West, Cameron; Crews, Bridgit; Mikel, Charles; Almazan, Perla; Latyshev, Sergey (2010-06-01). "An evaluation of the diagnostic accuracy of liquid chromatography-tandem mass spectrometry versus immunoassay drug testing in pain patients". Pain Physician. 13 (3): 273–281. PMID 20495592.
- ↑ Tsai, I.-Lin; Weng, Te-I.; Tseng, Yufeng J.; Tan, Happy Kuy-Lok; Sun, Hsiao-Ju; Kuo, Ching-Hua (2013-12-01). "Screening and confirmation of 62 drugs of abuse and metabolites in urine by ultra-high-performance liquid chromatography-quadrupole time-of-flight mass spectrometry". Journal of Analytical Toxicology. 37 (9): 642–651. PMID 24084874. doi:10.1093/jat/bkt083.
- ↑ Weinmann, W.; Renz, M.; Vogt, S.; Pollak, S. (2000-01-01). "Automated solid-phase extraction and two-step derivatisation for simultaneous analysis of basic illicit drugs in serum by GC/MS". International Journal of Legal Medicine. 113 (4): 229–235. PMID 10929239. doi:10.1007/s004149900098.
- ↑ Kolmonen, Marjo; Leinonen, Antti; Pelander, Anna; Ojanperä, Ilkka (2007-02-28). "A general screening method for doping agents in human urine by solid phase extraction and liquid chromatography/time-of-flight mass spectrometry". Analytica Chimica Acta. 585 (1): 94–102. PMID 17386652. doi:10.1016/j.aca.2006.12.028.
- ↑ Pelander, Anna; Ojanperä, Ilkka; Laks, Suvi; Rasanen, Ilpo; Vuori, Erkki (2003-11-01). "Toxicological screening with formula-based metabolite identification by liquid chromatography/time-of-flight mass spectrometry". Analytical Chemistry. 75 (21): 5710–5718. PMID 14588010. doi:10.1021/ac030162o.
- ↑ Nobilis, Milan; Pour, Milan; Senel, Petr; Pavlík, Jan; Kunes, Jirí; Voprsalová, Marie; Kolárová, Lenka; Holcapek, Michal (2007-06-15). "Metabolic profiling of a potential antifungal drug, 3-(4-bromophenyl)-5-acetoxymethyl-2,5-dihydrofuran-2-one, in mouse urine using high-performance liquid chromatography with UV photodiode-array and mass spectrometric detection". Journal of Chromatography B. 853 (1–2): 10–19. PMID 17400036. doi:10.1016/j.jchromb.2007.02.045.
- ↑ Sundström, Mira; Pelander, Anna; Angerer, Verena; Hutter, Melanie; Kneisel, Stefan; Ojanperä, Ilkka (2013-10-01). "A high-sensitivity ultra-high performance liquid chromatography/high-resolution time-of-flight mass spectrometry (UHPLC-HR-TOFMS) method for screening synthetic cannabinoids and other drugs of abuse in urine". Analytical and Bioanalytical Chemistry. 405 (26): 8463–8474. PMID 23954996. doi:10.1007/s00216-013-7272-8.
- ↑ Zahedi Rad, Maliheh; Neyestani, Tirang Reza; Nikooyeh, Bahareh; Shariatzadeh, Nastaran; Kalayi, Ali; Khalaji, Niloufar; Gharavi, Azam (2015-01-01). "Competitive Protein-binding assay-based Enzyme-immunoassay Method, Compared to High-pressure Liquid Chromatography, Has a Very Lower Diagnostic Value to Detect Vitamin D Deficiency in 9–12 Years Children". International Journal of Preventive Medicine. 6: 67. PMC 4542329 . PMID 26330983. doi:10.4103/2008-7802.161069.
Further reading
- L. R. Snyder, J.J. Kirkland, and J. W. Dolan, Introduction to Modern Liquid Chromatography, John Wiley & Sons, New York, 2009.
- M.W. Dong, Modern HPLC for practicing scientists. Wiley, 2006.
- L. R. Snyder, J.J. Kirkland, and J. L. Glajch, Practical HPLC Method Development, John Wiley & Sons, New York, 1997.
- S. Ahuja and H. T. Rasmussen (ed), HPLC Method Development for Pharmaceuticals, Academic Press, 2007.
- S. Ahuja and M.W. Dong (ed), Handbook of Pharmaceutical Analysis by HPLC, Elsevier/Academic Press, 2005.
- Y. V. Kazakevich and R. LoBrutto (ed.), HPLC for Pharmaceutical Scientists, Wiley, 2007.
- U. D. Neue, HPLC Columns: Theory, Technology, and Practice, Wiley-VCH, New York, 1997.
- M. C. McMaster, HPLC, a practical user's guide, Wiley, 2007.
External links
| Wikimedia Commons has media related to High performance liquid chromatography. |
| Look up HPLC in Wiktionary, the free dictionary. |