Thalassemia
| Thalassemia | |
|---|---|
| Synonyms | Thalassaemia, Mediterranean anemia |
 | |
| Peripheral blood film from a person with Delta Beta thalassemia | |
| Specialty | Hematology |
| Symptoms | Feeling tired, pale skin, enlarged spleen, yellowish skin, dark urine[1] |
| Causes | Genetic disorders (inherited)[2] |
| Diagnostic method | Blood tests, genetic tests[3] |
| Treatment | Blood transfusions, iron chelation, folic acid[4] |
| Frequency | 280 million (2015)[5] |
| Deaths | 16,800 (2015)[6] |
Thalassemias are inherited blood disorders characterized by abnormal hemoglobin production.[7] Symptoms depend on the type and can vary from none to severe.[1] Often there is mild to severe anemia (low red blood cells). Anemia can result in feeling tired and pale skin. There may also be bone problems, an enlarged spleen, yellowish skin, dark urine, and among children slow growth.[1]
Thalassemias are genetic disorders inherited from a person's parents.[2] There are two main types, alpha thalassemia and beta thalassemia.[7] The severity of alpha and beta thalassemia depends on how many of the four genes for alpha globin or two genes for beta globin are missing.[2] Diagnosis is typically by blood tests including a complete blood count, special hemoglobin tests, and genetic tests.[3] Diagnosis may occur before birth through prenatal testing.[8]
Treatment depends on the type and severity. Treatment for those with more severe disease often includes regular blood transfusions, iron chelation, and folic acid. Iron chelation may be done with deferoxamine or deferasirox. Occasionally, a bone marrow transplant may be an option.[4] Complications may include iron overload from the transfusions with resulting heart or liver disease, infections, and osteoporosis. If the spleen becomes overly enlarged, surgical removal may be required.[1]
As of 2013, thalassemia occurs in about 280 million people, with about 439,000 having severe disease.[9] It is most common among people of Italian, Greek, Middle Eastern, South Asian, and African descent.[7] Males and females have similar rates of disease.[10] It resulted in 16,800 deaths in 2015, down from 36,000 deaths in 1990.[11][6] Those who have minor degrees of thalassemia, similar to those with sickle-cell trait, have some protection against malaria, explaining why they are more common in regions of the world where malaria exists.[12]
Signs and symptoms
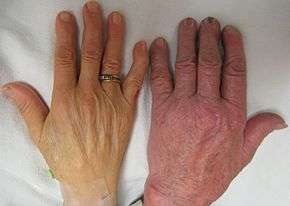
- Iron overload: People with thalassemia can get an overload of iron in their bodies, either from the disease itself or from frequent blood transfusions. Too much iron can result in damage to the heart, liver, and endocrine system, which includes glands that produce hormones that regulate processes throughout the body. The damage is characterized by excessive deposits of iron. Without adequate iron chelation therapy, almost all patients with beta-thalassemia accumulate potentially fatal iron levels.[13]
- Infection: People with thalassemia have an increased risk of infection. This is especially true if the spleen has been removed.[14]
- Bone deformities: Thalassemia can make the bone marrow expand, which causes bones to widen. This can result in abnormal bone structure, especially in the face and skull. Bone marrow expansion also makes bones thin and brittle, increasing the risk of broken bones.[15]
- Enlarged spleen: The spleen aids in fighting infection and filters unwanted material, such as old or damaged blood cells. Thalassemia is often accompanied by the destruction of a large number of red blood cells and the task of removing these cells causes the spleen to enlarge. Splenomegaly can make anemia worse, and it can reduce the life of transfused red blood cells. Severe enlargement of the spleen may necessitate its removal.[16]
- Slowed growth rates: anemia can cause a child's growth to slow. Puberty also may be delayed in children with thalassemia.[17]
- Heart problems: Diseases, such as congestive heart failure and abnormal heart rhythms, may be associated with severe thalassemia.[18]
Cause

Both α- and β-thalassemias are often inherited in an autosomal recessive manner. Cases of dominantly inherited α- and β-thalassemias have been reported, the first of which was in an Irish family with two deletions of 4 and 11 bp in exon 3 interrupted by an insertion of 5 bp in the β-globin gene. For the autosomal recessive forms of the disease, both parents must be carriers for a child to be affected. If both parents carry a hemoglobinopathy trait, the risk is 25% for each pregnancy for an affected child.
Estimates suggest that approximately 1.5% of the global population (80 - 90 million people) are β-thalassemia carriers.[19] However, exact data on carrier rates in many populations are lacking, particularly in developing areas of the world known or expected to be heavily affected.[20][21] Because of the prevalence of the disease in countries with little knowledge of thalassemia, access to proper treatment and diagnosis can be difficult.[22] While there are some diagnostic and treatment facilities in developing countries, in most cases these are not provided by government services, and are available only to patients that can afford them. In general, poorer populations only have access to limited diagnostic facilities together with blood transfusions. In some developing countries, there are virtually no facilities for diagnosis or management of thalassemia.[22]
Evolution
Having a single genetic variant for thalassemia may protect against malaria and thus be an advantage.[23]
People diagnosed with heterozygous (carrier) β-thalassemia have some protection against coronary heart disease.[24]
Pathophysiology
Normally, the majority of adult hemoglobin (HbA) is composed of four protein chains, two α and two β globin chains arranged into a heterotetramer. In thalassemia, patients have defects in either the α or β globin chain, causing production of abnormal red blood cells (In sickle-cell disease, the mutation is specific to β globin).
The thalassemias are classified according to which chain of the hemoglobin molecule is affected. In α-thalassemias, production of the α globin chain is affected, while in β-thalassemia, production of the β globin chain is affected.
The β globin chains are encoded by a single gene on chromosome 11; α globin chains are encoded by two closely linked genes on chromosome 16.[25] Thus, in a normal person with two copies of each chromosome, two loci encode the β chain, and four loci encode the α chain. Deletion of one of the α loci has a high prevalence in people of African or Asian descent, making them more likely to develop α-thalassemia. β-Thalassemias are not only common in Africans, but also in Greeks and Italians.
Alpha-thalassemias
The α-thalassemias involve the genes HBA1[26] and HBA2,[27] inherited in a Mendelian recessive fashion. Two gene loci and so four alleles exist. It is also connected to the deletion of the 16p chromosome. α Thalassemias result in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers (called hemoglobin H or HbH of 4 beta chains), which have abnormal oxygen dissociation curves.
Beta-thalassemia
Beta thalassemias are due to mutations in the HBB gene on chromosome 11,[28] also inherited in an autosomal, recessive fashion. The severity of the disease depends on the nature of the mutation and on the presence of mutations in one or both alleles.
Mutated alleles are called β+ when partial function is conserved (either the protein has a reduced function, or it functions normally but is produced in reduced quantity) or βo, when no functioning protein is produced.
The situation of both alleles determines the clinical picture:
- β thalassemia major (Mediterranean anemia or Cooley anemia) is caused by a βo/βo genotype. No functional β chains are produced, and thus no hemoglobin A can be assembled. This is the most severe form of β-thalassemia;
- β thalassemia intermedia is caused by a β+/βo or β+/β+ genotype. In this form, some hemoglobin A is produced;
- β thalassemia minor is caused by a β/βo or β/β+ genotype. Only one of the two β globin alleles contains a mutation, so β chain production is not terribly compromised and patients may be relatively asymptomatic.
Delta-thalassemia
As well as alpha and beta chains present in hemoglobin, about 3% of adult hemoglobin is made of alpha and delta chains. Just as with beta thalassemia, mutations that affect the ability of this gene to produce delta chains can occur.
Combination hemoglobinopathies
Thalassemia can coexist with other hemoglobinopathies. The most common of these are:
- Hemoglobin E/thalassemia: common in Cambodia, Thailand, and parts of India, it is clinically similar to β thalassemia major or thalassemia intermedia.
- Hemoglobin S/thalassemia: common in African and Mediterranean populations, is clinically similar to sickle-cell anemia, with the additional feature of splenomegaly.
- Hemoglobin C/thalassemia: common in Mediterranean and African populations, hemoglobin C/βo thalassemia causes a moderately severe hemolytic anemia with splenomegaly; hemoglobin C/β+ thalassemia produces a milder disease.
- Hemoglobin D/thalassemia: common in the northwestern parts of India and Pakistan (Punjab region).
Prevention
The American College of Obstetricians and Gynecologists recommends all people thinking of becoming pregnant be tested to see if they have thalassemia.[29] Genetic counseling and genetic testing are recommended for families who carry a thalassemia trait.
A screening policy exists in Cyprus to reduce the rate of thalassemia, which, since the program's implementation in the 1970s (which also includes prenatal screening and abortion), has reduced the number of children born with the disease from one of every 158 births to almost zero.[30]
In Iran as a premarital screening, the man's red cell indices are checked first, if he has microcytosis (mean cell hemoglobin < 27 pg or mean red cell volume < 80 fl), the woman is tested. When both are microcytic, their hemoglobin A2 concentrations are measured. If both have a concentration above 3.5% (diagnostic of thalassemia trait) they are referred to the local designated health post for genetic counseling.[31]
Large scale awareness campaigns are being organized in India[32] both by government and non-government organizations in favor of voluntary premarital screening to detect carriers of thalassemia and marriage between both carriers are strongly discouraged.
Management
Mild thalassemia: people with thalassemia traits do not require medical or follow-up care after the initial diagnosis is made.[33] People with β-thalassemia trait should be warned that their condition can be misdiagnosed as the more common iron deficiency anemia. They should avoid routine use of iron supplements; iron deficiency can develop, though, during pregnancy or from chronic bleeding.[34] Counseling is indicated in all persons with genetic disorders, especially when the family is at risk of a severe form of disease that may be prevented.[35]
Blood transfusions
People with severe thalassemia require medical treatment. A blood transfusion regimen was the first measure effective in prolonging life.[33]
Medications
Multiple blood transfusions can result in iron overload. The iron overload related to thalassemia may be treated by chelation therapy with the medications deferoxamine, deferiprone, or deferasirox.[36] These treatments have resulted in improving life expectancy in those with thalassemia major.[36]
Deferoxamine is only effective via daily injections which makes its long-term use more difficult. It has the benefit of being inexpensive and decent long-term safety. Adverse effects are primary skin reactions around the injection site and hearing loss.[36]
Deferasirox has the benefit of being an oral medication. Common side effects include: nausea, vomiting and diarrhea. It however is not effective in everyone and is probably not suitable in those with significant cardiac issues related to iron overload. The cost is also significant.[36]
Deferiprone is a medication that is given by mouth. Nausea, vomiting, and diarrhea are relatively common with its use.[36] It is available in both Europe and the United States.[36][37] It appears to be the most effective agent when the heart is significantly involved.[36]
There is no evidence from randomized controlled trial to support zinc supplementation in thalassemia.[38]
Bone marrow transplant
Bone marrow transplantation may offer the possibility of a cure in young people who have an HLA-matched donor.[39] Success rates have been in the 80–90% range.[39] Mortality from the procedure is about 3%.[40] There are no randomized controlled trials which have tested the safety and efficacy of non-identical donor bone marrow transplantation in persons with β- thalassemia who are dependent on blood transfusion.[41]
If the person does not have an HLA-matched compatible donor, another method called bone marrow transplantation (BMT) from haploidentical mother to child (mismatched donor) may be used. In a study of 31 people, the thalassemia-free survival rate 70%, rejection 23%, and mortality 7%. The best results are with very young people.[42]
Epidemiology
The beta form of thalassemia is particularly prevalent among Mediterranean peoples, and this geographical association is responsible for its naming.[43] Thalassemia resulted in 25,000 deaths in 2013 down from 36,000 deaths in 1990.[11]
In Europe, the highest concentrations of the disease are found in Greece, coastal regions in Turkey (particularly the Aegean Region such as Izmir, Balikesir, Aydin, Mugla, and Mediterranean Region such as Antalya, Adana, Mersin), in parts of Italy, particularly southern Italy and the lower Po valley. The major Mediterranean islands (except the Balearics) such as Sicily, Sardinia, Malta, Corsica, Cyprus, and Crete are heavily affected in particular. Other Mediterranean people, as well as those in the vicinity of the Mediterranean, also have high rates of thalassemia, including people from West Asia and North Africa. Far from the Mediterranean, South Asians are also affected, with the world's highest concentration of carriers (30% of the population) being in the Maldives.
Nowadays, it is found in populations living in Africa, the Americas, and in Tharu people in the Terai region of Nepal and India.[44] It is believed to account for much lower malaria sicknesses and deaths,[45] accounting for the historic ability of Tharus to survive in areas with heavy malaria infestation, where others could not. Thalassemias are particularly associated with people of Mediterranean origin, Arabs (especially Palestinians and people of Palestinian descent), and Asians.[46] The Maldives has the highest incidence of thalassemia in the world with a carrier rate of 18% of the population. The estimated prevalence is 16% in people from Cyprus, 1%[47] in Thailand, and 3–8% in populations from Bangladesh, China, India, Malaysia and Pakistan. Thalassemias also occur in descendants of people from Mediterranean countries (e.g. Greece, Italy, Spain, and others), in Latin America.
Etymology and pronunciation
The word thalassemia (/θælɪˈsiːmiə/) derives from the Greek thalassa (θάλασσα), "sea", and New Latin -emia (from Greek haema (αἷμα), "blood"). It was coined because the condition called "Mediterranean anemia" was first described in people of Mediterranean ethnicities. "Mediterranean anemia" was renamed thalassemia major once the genetics were better understood. The word thalassemia was first used in 1932.[43]:877[48]
Society and culture
In 2008, in Spain, a baby was selectively implanted to be a cure for his brother's thalassemia. The child was born from an embryo screened to be free of the disease before implantation with in vitro fertilization. The baby's supply of immunologically compatible cord blood was saved for transplantation to his brother. The transplantation was considered successful.[49] In 2009, a group of doctors and specialists in Chennai and Coimbatore registered the successful treatment of thalassemia in a child using an unaffected sibling's umbilical cord blood.[50]
References
- 1 2 3 4 "What Are the Signs and Symptoms of Thalassemias?". NHLBI. July 3, 2012. Retrieved 5 September 2016.
- 1 2 3 "What Causes Thalassemias?". NHLBI. July 3, 2012. Retrieved 5 September 2016.
- 1 2 "How Are Thalassemias Diagnosed?". NHLBI. July 3, 2012. Retrieved 5 September 2016.
- 1 2 "How Are Thalassemias Treated?". NHLBI. July 3, 2012. Retrieved 5 September 2016.
- ↑ GBD 2015 Disease and Injury Incidence and Prevalence, Collaborators. (8 October 2016). "Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015.". Lancet. 388 (10053): 1545–1602. PMID 27733282.
- 1 2 GBD 2015 Mortality and Causes of Death, Collaborators. (8 October 2016). "Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015.". Lancet. 388 (10053): 1459–1544. PMID 27733281.
- 1 2 3 "What Are Thalassemias?". NHLBI. July 3, 2012. Retrieved 5 September 2016.
- ↑ "How Can Thalassemias Be Prevented?". NHLBI. July 3, 2012. Retrieved 5 September 2016.
- ↑ Global Burden of Disease Study 2013, Collaborators (22 August 2015). "Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013.". Lancet. 386 (9995): 743–800. PMC 4561509
 . PMID 26063472. doi:10.1016/s0140-6736(15)60692-4.
. PMID 26063472. doi:10.1016/s0140-6736(15)60692-4. - ↑ Clin. Methods in Ped. Jaypee Brothers Publishers. 2005. p. 21. ISBN 9788171798087.
- 1 2 GBD 2013 Mortality and Causes of Death, Collaborators (17 December 2014). "Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013.". Lancet. 385: 117–71. PMC 4340604 . PMID 25530442. doi:10.1016/S0140-6736(14)61682-2.
- ↑ Weatherall, D. J. (2015). "The Thalassemias: Disorders of Globin Synthesis". Williams Hematology (9e ed.). McGraw Hill Professional. p. 725. ISBN 9780071833011.
- ↑ Cianciulli P (October 2008). "Treatment of iron overload in thalassemia". Pediatr Endocrinol Rev. 6 (Suppl 1): 208–13. PMID 19337180.
- ↑ http://www.mayoclinic.org/diseases-conditions/thalassemia/symptoms-causes/dxc-20261829
- ↑ Vogiatzi, Maria G; Macklin, Eric A; Fung, Ellen B; Cheung, Angela M; Vichinsky, Elliot; Olivieri, Nancy; Kirby, Melanie; Kwiatkowski, Janet L; Cunningham, Melody; Holm, Ingrid A; Lane, Joseph; Schneider, Robert; Fleisher, Martin; Grady, Robert W; Peterson, Charles C; Giardina, Patricia J (March 2009). "Bone Disease in Thalassemia: A Frequent and Still Unresolved Problem". Journal of Bone and Mineral Research. 24 (3): 543–557. ISSN 0884-0431. PMC 3276604 . PMID 18505376. doi:10.1359/jbmr.080505.
- ↑ "Symptoms and causes - Enlarged spleen (splenomegaly) - Mayo Clinic". www.mayoclinic.org. Retrieved 2017-02-02.
- ↑ Soliman, Ashraf T; Kalra, Sanjay; De Sanctis, Vincenzo (2014-11-01). "Anemia and growth". Indian Journal of Endocrinology and Metabolism. 18 (7). PMC 4266864 . PMID 25538873. doi:10.4103/2230-8210.145038.
- ↑ "Thalassemia Complications". Thalassemia. Open Publishing. Retrieved 27 September 2011.
- ↑ Galanello, Renzo; Origa, Raffaella (2010). "Beta-thalassemia". Orphanet Journal of Rare Diseases. 5 (1): 11. PMC 2893117 . doi:10.1186/1750-1172-5-11.
- ↑ Galanello, Renzo; Origa, Raffaella (2010). "Beta-thalassemia". Orphanet Journal of Rare Diseases. 5 (1): 11. doi:10.1186/1750-1172-5-11.
- ↑ Vichinsky, Elliott P. (1 November 2005). "Changing Patterns of Thalassemia Worldwide". Annals of the New York Academy of Sciences. 1054 (1): 18–24. ISSN 1749-6632. doi:10.1196/annals.1345.003.
- 1 2 WEATHERALL, DAVID J. (November 2005). "Keynote Address: The Challenge of Thalassemia for the Developing Countries". Annals of the New York Academy of Sciences. 1054 (1): 11–17. doi:10.1196/annals.1345.002.
- ↑ Wambua S; Mwangi, Tabitha W.; Kortok, Moses; Uyoga, Sophie M.; Macharia, Alex W.; Mwacharo, Jedidah K.; Weatherall, David J.; Snow, Robert W.; Marsh, Kevin; Williams, Thomas N. (May 2006). "The Effect of α +-Thalassaemia on the Incidence of Malaria and Other Diseases in Children Living on the Coast of Kenya". PLoS Medicine. 3 (5): e158. PMC 1435778 . PMID 16605300. doi:10.1371/journal.pmed.0030158.
- ↑ Tassiopoulos S; Deftereos, Spyros; Konstantopoulos, Kostas; Farmakis, Dimitris; Tsironi, Maria; Kyriakidis, Michalis; Aessopos, Athanassios (2005). "Does heterozygous beta-thalassemia confer a protection against coronary artery disease?". Annals of the New York Academy of Sciences. 1054: 467–70. PMID 16339699. doi:10.1196/annals.1345.068.
- ↑ Robbins Basic Pathology, Page No:428
- ↑ Online Mendelian Inheritance in Man (OMIM) Hemoglobin—Alpha locus 1; HBA1 -141800
- ↑ Online Mendelian Inheritance in Man (OMIM) Hemoglobin—Alpha locus 2; HBA2 -141850
- ↑ Online Mendelian Inheritance in Man (OMIM) Hemoglobin—Beta Locus; HBB -141900
- ↑ "Carrier Screening in the Age of Genomic Medicine - ACOG". www.acog.org. Retrieved 24 February 2017.
- ↑ Leung TN; Lau TK; Chung TKh (April 2005). "Thalassaemia screening in pregnancy". Current Opinion in Obstetrics and Gynecology. 17 (2): 129–34. PMID 15758603. doi:10.1097/01.gco.0000162180.22984.a3.
- ↑ Samavat A, Modell B (November 2004). "Iranian national thalassaemia screening programme". BMJ (Clinical Research Ed.). 329 (7475): 1134–7. PMC 527686 . PMID 15539666. doi:10.1136/bmj.329.7475.1134.
- ↑ Petrou, Mary (2010-01-01). "Screening for beta thalassaemia". Indian Journal of Human Genetics. 16 (1). PMC 2927788 . PMID 20838484. doi:10.4103/0971-6866.64934.
- 1 2 Pediatric Thalassemia~treatment at eMedicine
- ↑ Burdick CO; Ntaios, G.; Rathod, D. (March 2009). "Separating thalassemia trait and iron deficiency by simple inspection". Am. J. Clin. Pathol. 131 (3): 444; author reply 445. PMID 19228649. doi:10.1309/AJCPC09VRAXEASMH.
- ↑ Harrison's Principles of Internal Medicine (17th ed.). McGraw-Hill medical. September 2008. p. 776. ISBN 0-07-164114-9.
- 1 2 3 4 5 6 7 Neufeld, EJ (2010). "Update on iron chelators in thalassemia.". Hematology / the Education Program of the American Society of Hematology. American Society of Hematology. Education Program. 2010: 451–5. PMID 21239834. doi:10.1182/asheducation-2010.1.451.
- ↑ "Ferriprox". Drugs.com. American Society of Health-System Pharmacists. Retrieved 5 September 2016.
- ↑ Kye Mon Min Swe (2013). "Zinc supplements for treating thalassaemia and sickle cell disease". Cochrane Database of Systematic Reviews (6): Art. No.: CD009415. PMID 23807756. doi:10.1002/14651858.CD009415.pub2.
- 1 2 Gaziev, J; Lucarelli, G (June 2011). "Hematopoietic stem cell transplantation for thalassemia.". Current Stem Cell Research & Therapy. 6 (2): 162–9. PMID 21190532. doi:10.2174/157488811795495413.
- ↑ Sabloff, M; Chandy, M; Wang, Z; Logan, BR; Ghavamzadeh, A; Li, CK; Irfan, SM; Bredeson, CN; et al. (2011). "HLA-matched sibling bone marrow transplantation for β-thalassemia major". Blood. 117 (5): 1745–50. PMC 3056598 . PMID 21119108. doi:10.1182/blood-2010-09-306829.
- ↑ Jagannath, Vanitha A.; Fedorowicz, Zbys; Al Hajeri, Amani; Sharma, Akshay (2016-11-30). "Hematopoietic stem cell transplantation for people with ß-thalassaemia major". The Cochrane Database of Systematic Reviews. 11: CD008708. ISSN 1469-493X. PMID 27900772. doi:10.1002/14651858.CD008708.pub4.
- ↑ Sodani, P; Isgrò, A; Gaziev, J; Paciaroni, K; Marziali, M; Simone, MD; Roveda, A; De Angelis, G; et al. (2011). "T cell-depleted hla-haploidentical stem cell transplantation in thalassemia young patients". Pediatric reports. 3 (Suppl 2): e13. PMC 3206538 . PMID 22053275. doi:10.4081/pr.2011.s2.e13.
- 1 2 John P. Greer JP, Arber DA, Glader B, et al. Wintrobe's Clinical Hematology 2013. ISBN 9781451172683
- ↑ Modiano, G.; Morpurgo, G; Terrenato, L; Novelletto, A; Di Rienzo, A; Colombo, B; Purpura, M; Mariani, M; et al. (1991). "Protection against malaria morbidity: Near-fixation of the α-thalassemia gene in a Nepalese population". American Journal of Human Genetics. 48 (2): 390–7. PMC 1683029 . PMID 1990845.
- ↑ Terrenato, L; Shrestha, S; Dixit, KA; Luzzatto, L; Modiano, G; Morpurgo, G; Arese, P (February 1988). "Decreased malaria morbidity in the Tharu people compared to sympatric populations in Nepal.". Annals of tropical medicine and parasitology. 82 (1): 1–11. PMID 3041928.
- ↑ E. Goljan, Pathology, 2nd ed. Mosby Elsevier, Rapid Review Series.
- ↑ "Thalassemia" (in Thai). Department of Medical Sciences,. September 2011. Archived from the original on 2011-09-25.
- ↑ Whipple GH, Bradford WI. Am J Dis Child 1932;44:336
- ↑ Spanish Baby Engineered To Cure Brother
- ↑ His sister's keeper: Brother's blood is boon of life, Times of India, 17 September 2009
External links
| Classification |
V · T · D |
|---|---|
| External resources |
- Thalassemia at DMOZ
- Learning About Thalassemia published by the National Human Genome Research Institute.