Hemoglobin Hopkins-2
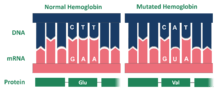
Hemoglobin Hopkins-2 (Hb Hop-2) is a mutation of the protein hemoglobin, which is responsible for the transportation of oxygen through the blood from the lungs to the musculature of the body in vertebrates. Generally, the mutation causes two abnormal α chains in the protein's structure.[1] Within the chains, the mutation is the result of hemoglobin's histidine amino acid being replaced with aspartic acid in the protein's genetic sequence.[1] This amino acid structure change occurs at residue 112. Additionally, within one of the mutated alpha chains, there are substitutes at 114 and 118, two points on the amino acid chain.[2] This mutation can cause sickle cell anemia.[3]
Following the initial discovery of hemoglobin, two researchers working at Johns Hopkins Hospital in the mid-twentieth century, Ernest W. Smith and J.V. Torbert, discovered the Hopkins-2 mutation of hemoglobin.[4] Work by Harvey A. Itano and Elizabeth A. Robinson in 1960 confirmed Smith's and Torbert's finding and emphasized the importance of the alpha loci in the mutation.[5] Later in the twentieth century, Samuel Charache, another Hopkins affiliated scientist and doctor, studied the physiological impacts of the variant on health.[6] His findings suggest that the variant plays no effect clinically.[7]
History
In the mid-1900s, many factors prompted hemoglobin research in Baltimore, Maryland and ultimately led to the discovery of Hemoglobin Hopkins-2. First, the development of new technology, including x-ray crystallography and protein chemistry, that could be utilized in molecular biology studies catalyzed research.[8] Furthermore, the large presence of thalassemia (Hb H), a disorder in which the alpha gene is dysfunctional, in Southeast Asia and southern China further concerned researchers as, if left untreated, the mutation could result in bone deformities, swelling of the spleen, slowed growth rate, or cardiac dysfunction.[9][10] Additionally, Max Perutz's, a Cambridge researcher, discovery of hemoglobin's basic tertiary structure in 1962 catalyzed research in hematology.[11] Vernon Ingram's research surrounding sickle cell anemia in 1956 revealed that variants, or mutations, in hemoglobin's RNA resulted in the sickle cell disease.[8][3]

Ernest W. Smith and J.V. Norbert examined Ingram's discovery and, in 1958, they discovered the hemoglobin Hopkins-2 mutation. Smith and Torbert, research fellows working in Lockard Conley's Hematology Research Department at Johns Hopkins University, proved that recombination of non-allelic genes resulted in two variants of the gene that produced hemoglobin. Variants of the "normal" hemoglobin gene result in mutation in the hemoglobins produced.[4] In other words, the researchers discovered two versions of hemoglobin: the [Hb-Hop2] and S variants.[8]
Smith's and Torbert's findings were confirmed and built upon by Harvey A. Itano and Elizabeth A. Robinson in 1960. In their paper, Genetic Control of the a- and B-Chains of Hemoglobin, Itano and Robinson explained that both the alpha and beta loci are involved in the regulation of hemoglobin. The recombination of the alpha loci on the gene coding for hemoglobin results in a mutation of the hemoglobin protein. Researchers refer to this mutated protein as a Hopkins-2 variation of hemoglobin.[5]
Further research surrounding the Hopkins-2 genetic mutation was conducted by Johns Hopkins doctors, who remained engaged in research in the hematologic field despite a general change in focus away from hematology in the medical community. Prominent Hopkins researcher Samuel Charache was one of many scientists at Hopkins who investigated blood and its components during the late twentieth century.[8] Specifically, Charache is well known for his work with George Dover, another Hopkins researcher; together, they discovered a treatment option for sickle cell anemia. However, Charache was also engaged in hemoglobin Hopkins-2 research. In his Nature article, titled Clinical Studies and Physiological Properties of Hopkins Hemoglobin-2, Charache announced his discoveries surrounding the hemoglobin variant. Charache focused on physical implications of the variant, rather than on genetics themselves. Ultimately, Charache asserts in his paper that the variant is not prominent phenotypically and plays an unnoticeable or no effect on human health.
Hemoglobin and Hematology at Hopkins

Hematologic studies were prominent at Johns Hopkins Hospital prior to the discovery of the hemoglobin Hopkins-2 variant. William Osler, one of the founders of Johns Hopkins Hospital, is credited with the discovery of platelets as the third component of blood, in addition to red blood cells and leukocytes. After observing the cells under a microscope, Osler connected the concept of dysfunctional platelets to the development of ulcerative endocarditis and thrombosis. Osler's initial observations of platelets catalyzed the study of blood and hematology at Hopkins and in other research environments.[12]
Beginning in 1920, doctors at Johns Hopkins Hospital conducted research on sickle cell anemia, or sickle cell disease. Although their conclusions surrounding the disease are outdated, Doctors Taliaferro and Huck discovered a latent form of sickle cell anemia. Their study on sickle cell anemia was the first of many to occur at Hopkins. In 1940, Irving Sherman, a medical student at Johns Hopkins, correctly identified the deoxygenation of hemoglobin in sickle cell patients after he noted refraction patterns characteristic of deoxygenation when light was passed through the protein.[13] The deoxygenation of hemoglobin in sickle cell patients has severe implications on those who carry the mutation. The hemoglobin proteins, present in those with sickle cell disease, cannot carry oxygen to the organs and other tissues of the human body. This results in pain crises and the disease results in an abbreviated life expectancy of 40–60 years.[14]
Ernest W. Smith and Torbert were integral in the discovery of Hopkins Hemoglobin-2, in addition to many other hematologic mutations and conditions.[4] The two scientists worked together at Hopkins to identify the N-Baltimore mutation of Hemoglobin in 1958.[15] Also referred to as the Hopkins-I, Jenkins, N-Memphis, or Kenwood mutation, the N-Baltimore mutation is a point mutation in which a glycine codon is replaced with an adenosine codon. The N-Baltimore mutation is associated with the C and S mutations of hemoglobin.[16]
Smith conducted extensive research in conjunction with Locklard Conley, one of Smith's bosses at the time of the Hopkins Hemoglobin-2 variant discovery. Lockard Conley, commonly referred to as "Lock," was a Johns Hopkins undergraduate and Columbia trained doctor. In 1947, Conley became the first director of the Hematology Department at Hopkins and remained in the position for 33 years. While there, he studied blood-related diseases, such as blood coagulation and sickle cell anemia, and invented machinery to analyze molecular species.[17] Specifically, he and Smith created a device that allowed for the separation of hemoglobin variants from standard hemoglobin molecules. Conley's impact on hematology, therefore, was not only scientific discovery; but, also, technological discovery that allowed the hematologic field to expand.[18] Conley remained a doctor and professor at Johns Hopkins Hospital until his death in 2010.[17]
Although a significant amount of hematologic research was completed during the 1950s and 1960s, scientists questioned whether more research could be completed without the development of more advanced technology. Ultimately, this doubt resulted in fewer scientists pursuing research in hematology in the mid twentieth century.[8] Despite the scarcity of researchers, new discoveries surrounding genetics and hemoglobin were made. However, research continued in major medical laboratories, like Hopkins.[19]
In the mid and late twentieth century, both doctors George J. Dover and Samuel Charache studied sickle cell anemia's pathology at Johns Hopkins Hospital.[7] Together, they implemented treatment of the disease through the use of a cancer drug, hydroxyurea; the drug was successful in alleviating some of the painful spurts associated with sickle cell anemia, in addition to pulmonary symptoms associated with the disease. Dover, a pediatric hematologist and expert on fetal hemoglobin, initiated the use of the protein as a way to treat sickle cell anemia in adults.
Medical Implications
The Hopkins-2 variant of hemoglobin has an oxygen affinity within the body, meaning that there is an increase of oxygen spreading though the body due to the fact that Ho-2 carries a higher amount of oxygen.[20] There is no red cell effect with Ho-2 compared to Hemoglobin S, which changes the shape of the cell to become sickled. The Hopkins-2 variant of hemoglobin is not involved in forming sickle cells. There is a lack of phenotypic expression of Ho-2 in terms of sickle cell, so a person with sickle cell and hemoglobin Hopkins-2 would be asymptomatic.
Genetic Basis
The Hopkins 2 variant of hemoglobin is the result of a mutation. Specifically, the mutated protein is composed of two alpha chains. The mutation for the Hopkins-2 variant of hemoglobin is located on the surface of the molecule. Within the Ho-2 variant, aspartic acid replaces histidine at position 112 on the alpha chain.[2] Dr. Max Perutz examined the aspartic acid in alpha 112 claiming that the carboxyl group of the amino acid forms a hydrogen bond with phenolix hydroxyl, which in turn stabilizes the structure of the molecule and increases oxygen affinity. Ho-2 hemoglobin has high levels of oxygen, which replaces the lack of oxygen within red blood cells. Ho-2 is similar to Hemoglobin A, which is normal hemoglobin and also contains two α-chains. This provides an explanation as to how the Hopkins-2 variant of hemoglobin is related to the symptoms of sickle cell.
Hopkins-2 can also interact specifically with Hemoglobin S. Hemoglobin S is the most common abnormal hemoglobin variant. Hemoglobin S is the variant that causes sickle cell, which is a disorder in which red blood cells break down and become abnormally shaped. Hemoglobin S has two beta chains, whereas hemoglobin Hopkins-2 has two alpha chains. Hopkins-2 makes up 22% of hemolysates in single heterozygotes; therefore, there is the normal version of the gene in these patients. Hemolysates are the products of the destruction of red blood cells. Ho-2 also comprises 11% of hemolysates in ‘double’ heterozygotes, which are when the gene contains both Hopkins-2 and Hemoglobin S.[21]
There are currently multiple possible explanations as to how the Hemoglobin variant Hopkins-2 works. One explanation that scientists have come up with is that Ho-2 is created due to a point mutation causing the substitution of histidine by aspartic acid.[21] The other explanation is that there is an unequal crossing over between two α genes which explains the replacement of histidine by aspartic acid.[21] This then led a deletion occurring within the chromosome housing Ho-2. This deletion removes the region where the N-terminus is located in the major α-chain and where the c-terminus is located on the minor α-chain.[21] Deletion of the N-terminus inactivates enzymes and halting their ability to cut chains at certain areas, which inevitably damages the chain.
Case Studies
Case studies were completed on some patients who carried the Hemoglobin Hopkins-2 genetic mutation by Samuel Charache and others. Researchers concluded after completing the studies that many of carriers of the mutation were asymptomatic to sickle cell and were overall quite healthy. Any medical issues that occurred had no correlation to sickle cell. These studies occurred in the 1970s.[8]
Fuller-Carr Family
There were five carriers of Hemoglobin Hopkins 2 in the Fuller-Carr family and ten double heterozygotes of Ho-2 and Hemoglobin S.[22] All the carriers were in good health and had normal hematology test results. Out of those carrying hemoglobin S and Ho-2, none were anemic; but, a few of those studied displayed elevated reticulocyte counts.[22] This is measured through a blood test that analyzes the speed of production of red blood cells by bone marrow and its release into the blood. There was no suggestion of symptomatic sickle cell anemia in the family.
Unknown Child
There was a study on a three year old that was a carrier of the hemoglobin variant of Hopkins-2. The child had mild anemia and reticulocytosis, which is commonly seen in anemia.[23] There were, however, no sickled cells found in the blood and they had no symptoms relating to sickle cell. There was also a reduced mean corpuscular volume (MCV), which is the average volume of red blood cell count.[23]
References
- 1 2 Clegg, J. B.; Charache, S. (1978-01-01). "The Structure of Hemoglobin Hopkins-2". Hemoglobin. 2 (1): 85–88. ISSN 0363-0269. doi:10.3109/03630267808999194.
- 1 2 Rucknagel, D. L.; Winter, W. P. (1974-11-01). "DUPLICATION OF STRUCTURAL GENES FOR HEMOGLOBIN α AND β CHAINS IN MAN *". Annals of the New York Academy of Sciences. 241 (1): 80–92. ISSN 1749-6632. doi:10.1111/j.1749-6632.1974.tb21868.x.
- 1 2 Bloom, Miriam (1995). Understanding Sickle Cell Disease. United States: University Press of Mississippi. pp. 3–6. ISBN 978-0878057450.
- 1 2 3 Hill, Robert L.; Swenson, Robert T.; Schwartz, Herbert C. (1960). "Characterization of a Chemical Abnormality in Hemoglobin G" (PDF). The Journal of Biological Chemistry. 235: 3182–3187.
- 1 2 Itano, Harvey A.; Robinson, Elizabeth A. (1960). "Genetic Control of the a- and B-Chains of Hemoglobin" (PDF). Proceedings of the National Academy of Sciences. 46: 1492–1501.
- ↑ Sugg, Diana K. (January 31, 1995). "Hopkins Finds Sickle Cell Treatment". The Baltimore Sun. Retrieved March 8, 2017.
- 1 2 Charache, Samuel (1971). "Clinical Studies and Physiological Properties of Hopkins-2 Haemoglobin" (PDF). Nature. 234: 248–251.
- 1 2 3 4 5 6 Weatherall, D.J. (2001). "Towards Molecular Medicine; Reminisces of the Haemoglobin Field" (PDF). British Journal of Haematology. 115: 729–738.
- ↑ "What is Thalassemia?". UCSF Benioff Children's Hospital. Retrieved March 8, 2017.
- ↑ "Symptoms and causes - Mayo Clinic". Mayo Clinic. Retrieved 2017-04-15.
- ↑ Steensma, David P.; Shampoo, Marc A.; Kyle, Robert A. (2015). "Max Perutz and the Structure of Hemoglobin". Mayo Clinic. 90: e89.
- ↑ Stone, Marvin J. (2003-10-01). "William Osler's Legacy and his Contribution to Haematology". British Journal of Haematology. 123 (1): 3–18. ISSN 1365-2141. doi:10.1046/j.1365-2141.2003.04615.x.
- ↑ "Discovery and Biological Basis". www.nslc.wustl.edu. Retrieved 2017-04-15.
- ↑ "What Is Sickle Cell Disease? - NHLBI, NIH". www.nhlbi.nih.gov. Retrieved 2017-04-20.
- ↑ Lorenzo-Medina, Mercedes; De-La-Iglesia, Silvia; Ropero, Paloma; Nogueira-Salgueiro, Patricia; Santana-Benitez, Jesus (2017-03-30). "Effects of Hemoglobin Variants on Hemoglobin A1c Values Measured Using a High-Performance Liquid Chromatography Method". Journal of Diabetes Science and Technology. 8 (6): 1168–1176. ISSN 1932-2968. PMC 4455477
 . PMID 25355712. doi:10.1177/1932296814538774.
. PMID 25355712. doi:10.1177/1932296814538774. - ↑ "N-Baltimore.html". globin.bx.psu.edu. Retrieved 2017-03-30.
- 1 2 "Medical Archives - Personal Paper Collections: The C. Lockard Conley Collection". www.medicalarchives.jhmi.edu. Retrieved 2017-03-30.
- ↑ "C. Lockard Lock Conley, MD (1915-2010)". www.hematology.org. 2010-04-27. Retrieved 2017-03-30.
- ↑ McCann, Shaun (2016). A History of Haematology: From Herodotus to HIV. Oxford University Press. ISBN 9780191027130.
- ↑ Huisman, Titus H. J, Marianne F. H. Carver, and Georgi Efremov. A Syllabus of Human Hemoglobin Variants (1996). Augusta, GA, USA: The Sickle Cell Anemia Foundation, 1996. Print.
- 1 2 3 4 Ostertag, W.; Von Ehrenstein, G.; Charache, S. (1972-05-17). "Duplicated α-Chain Genes in Hopkins-2 Haemoglobin of Man and Evidence for Unequal Crossing Over between Them". Nature. 237 (72): 90–94. doi:10.1038/10.1038/newbio237090a0.
- 1 2 Charache, S.; Ostertag, W.; Von Ehrenstein, G. (1972-05-17). "Clinical Studies and Physiological Properties of Hopkins-2 Haemoglobin". Nature. 237 (72): 88–90. doi:10.1038/10.1038/newbio237088a0.
- 1 2 Steinberg, Martin H., et al. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Cambridge University Press, 2009.