Grignard reaction
| Grignard reaction | |
|---|---|
| Named after | Victor Grignard |
| Reaction type | Coupling reaction |
| Identifiers | |
| Organic Chemistry Portal | grignard-reaction |
| RSC ontology ID | RXNO:0000014 |

The Grignard reaction (pronounced /ɡriɲar/) is an organometallic chemical reaction in which alkyl, vinyl, or aryl-magnesium halides (Grignard reagents) add to a carbonyl group in an aldehyde or ketone.[1][2] This reaction is an important tool for the formation of carbon–carbon bonds.[3][4] The reaction of an organic halide with magnesium is not a Grignard reaction, but provides a Grignard reagent.[5]
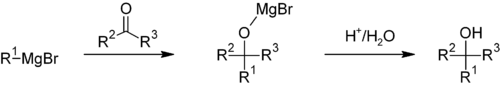
Grignard reactions and reagents were discovered by and are named after the French chemist François Auguste Victor Grignard (University of Nancy, France), who published it in 1900 and was awarded the 1912 Nobel Prize in Chemistry for this work.[6] Grignard reagents are similar to organolithium reagents because both are strong nucleophiles that can form new carbon–carbon bonds. The nucleophilicity increases if the alkyl substituent is replaced by an amido group. These amido magnesium halides are called Hauser bases.
Reaction mechanism
The Grignard reagent functions as a nucleophile, attacking the electrophilic carbon atom that is present within the polar bond of a carbonyl group. The addition of the Grignard reagent to the carbonyl typically proceeds through a six-membered ring transition state.[7]

However, with hindered Grignard reagents, the reaction may proceed by single-electron transfer. Similar pathways are assumed for other reactions of Grignard reagents, e.g., in the formation of carbon–phosphorus, carbon–tin, carbon–silicon, carbon–boron and other carbon–heteroatom bonds.
Preparation of Grignard reagent
Grignard reagents form via the reaction of an alkyl or aryl halide with magnesium metal. The reaction is conducted by adding the organic halide to a suspension of magnesium in an etherial solvent, which provides ligands required to stabilize the organomagnesium compound. Empirical evidence suggests that the reaction takes place on the surface of the metal. The reaction proceeds through single electron transfer:[8][9][10] In the Grignard formation reaction, radicals may be converted into carbanions through a second electron transfer.[11][12]
- R−X + Mg → R−X•− + Mg•+
- R−X•− → R• + X−
- R• + Mg•+ → RMg+
- RMg+ + X− → RMgX
A limitation of Grignard reagents is that they do not readily react with alkyl halides via an SN2 mechanism. On the other hand, they readily participate in transmetalation reactions:
- RMgX + AlX → AlR + MgX2
For this purpose, commercially available Grignard reagents are especially useful because this route avoids the problem with initiation.[13]
Reaction conditions
In reactions involving Grignard reagents, it is important to exclude water and air, which rapidly destroy the reagent by protonolysis or oxidation.[14] Since most Grignard reactions are conducted in anhydrous diethyl ether or tetrahydrofuran, side-reactions with air are limited by the protective blanket provided by solvent vapors. Small-scale or quantitative preparations should be conducted under nitrogen or argon atmospheres, using air-free techniques. Although the reagents still need to be dry, ultrasound can allow Grignard reagents to form in wet solvents by activating the magnesium such that it consumes the water.[15]
The organic halide
Grignard reactions often start slowly. As is common for reactions involving solids and solution, initiation follows an induction period during which reactive magnesium becomes exposed to the organic reagents. After this induction period, the reactions can be highly exothermic. Alkyl and aryl bromides and iodides are common, with chlorides being seen as well. However, fluorides are generally unreactive, except with specially activated magnesium (through Rieke metals).
Magnesium
Typical Grignard reactions involve the use of magnesium ribbon. All magnesium is coated with a passivating layer of magnesium oxide, which inhibits reactions with the organic halide. Specially activated magnesium, such as Rieke magnesium, circumvents this problem.[16] The oxide layer can also be broken up using ultrasound,[17] or by adding a few drops of iodine or 1,2-Diiodoethane.
Solvent

Most Grignard reactions are conducted in ethereal solvents, especially diethyl ether and THF. With the chelating diether dioxane, some Grignard reagents undergo a redistribution reaction to give diorganomagnesium compounds (R = organic group, X = halide):
- 2 RMgX + dioxane ⇌ R2Mg + MgX2(dioxane)
This reaction is known as the Schlenk equilibrium.
Testing Grignard reagents
Because Grignard reagents are so sensitive to moisture and oxygen, many methods have been developed to test the quality of a batch. Typical tests involve titrations with weighable, anhydrous protic reagents, e.g. menthol in the presence of a color-indicator. The interaction of the Grignard reagent with phenanthroline[18] or 2,2'-bipyridine causes a color change.
Initiation
Many methods have been developed to initiate sluggish Grignard reactions. These methods weaken the passivating layer of MgO, thereby exposing highly reactive magnesium to the organic halide. Mechanical methods include crushing of the Mg pieces in situ, rapid stirring, and sonication[19] of the suspension. Iodine, methyl iodide, and 1,2-Dibromoethane are common activating agents. The use of 1,2-dibromoethane is particularly advantageous as its action can be monitored by the observation of bubbles of ethylene. Furthermore, the side-products are innocuous:
- Mg + BrC2H4Br → C2H4 + MgBr2
The amount of Mg consumed by these activating agents is usually insignificant. A small amount of mercuric chloride will amalgamate the surface of the metal, allowing it to react.
Industrial production
Grignard reagents are produced in industry for use in situ, or for sale. As with a bench-scale, the main problem is that of initiation; a portion of a previous batch of Grignard reagent is often used as the initiator. Grignard reactions are exothermic, and this exothermicity must be considered when a reaction is scaled-up from laboratory to production plant.[20]
Many Grignard reagents such as methylmagnesium bromide, methylmagnesium chloride, phenylmagnesium bromide, and allylmagnesium bromide are available commercially as tetrahydrofuran or diethyl ether solutions.
Mg transfer reaction (halogen–Mg exchange)
An alternative preparation of Grignard reagents involves transfer of Mg from a preformed Grignard reagent to an organic halide. This method offers the advantage that the Mg transfer tolerates many functional groups. A typical reaction involves isopropylmagnesium chloride and aryl bromide or iodides.[21]
Reactions of Grignard reagents
With carbonyl compounds
Grignard reagents react with a variety of carbonyl derivatives.[22]
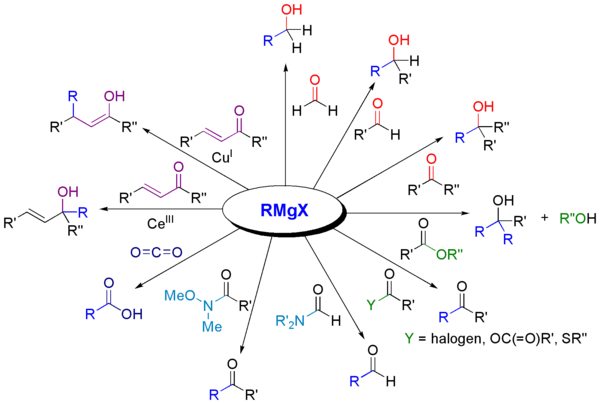
The most common application of Grignard reagents is the alkylation of aldehydes and ketones, i.e. the Grignard reaction:[23]

Note that the acetal function (a protected carbonyl) does not react.
Such reactions usually involve an aqueous acidic workup, though this step is rarely shown in reaction schemes. In cases where the Grignard reagent is adding to an aldehyde or a prochiral ketone, the Felkin-Anh model or Cram's Rule can usually predict which stereoisomer will be formed. With easily deprotonated 1,3-diketones and related acidic substrates, the Grignard reagent RMgX functions merely as a base, giving the enolate anion and liberating the alkane RH.
Grignard reagents are nucleophiles in nucleophilic aliphatic substitutions for instance with alkyl halides in a key step in industrial Naproxen production:

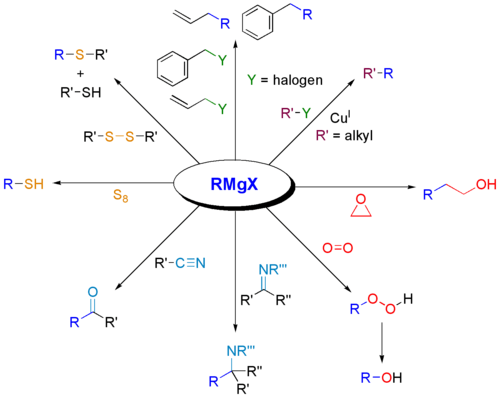
Reactions as a base
Grignard reagents serve as a base for protic substrates (this scheme does not show workup conditions, which typically includes water). Grignard reagents are basic and react with alcohols, phenols, etc. to give alkoxides (ROMgBr). The phenoxide derivative is susceptible to formylation paraformaldehyde to give salicylaldehyde.[24]
Formation of bonds to B, Si, P, Sn
Like organolithium compounds, Grignard reagents are useful for forming carbon–heteroatom bonds.
![{\displaystyle {\begin{matrix}{\ce {R4B-}}\\{\color {White}\scriptstyle {\ce {Et2O.BF3\ or\ NaBF4}}}{\Bigg \uparrow }\scriptstyle {\ce {Et2O.BF3\ or\ NaBF4}}\\{\ce {Ph2PR<-[{\ce {Ph2PCl}}]RMgX->[{\ce {Bu3SnCl}}]Bu3SnR}}\\{\color {White}\scriptstyle {\ce {B(OMe)3}}}{\Bigg \downarrow }\scriptstyle {\ce {B(OMe)3}}\\{\ce {RB(OMe)2}}\end{matrix}}}](../I/m/e437b60e37160b69a20e287dba0820ae949a6a4d.svg "Reactions of Grignard reagents with non carbon electrophiles")
Reaction with transition metal halides
Grignard reagents react with many metal-based electrophiles. For example, they undergo transmetallation with cadmium chloride (CdCl2) to give dialkylcadmium:[25]
- 2 RMgX + CdCl2 → R2Cd + 2 Mg(X)Cl
Dialkylcadmium reagents are used for preparation of ketones from acyl halides:
- 2 R'C(O)Cl + R2Cd → 2 R'C(O)R + CdCl2
With organic halides
Grignard reagents do not typically react with organic halides, in contrast with their high reactivity with other main group halides. In the presence of metal catalysts, however, Grignard reagents participate in C-C coupling reactions. For example, nonylmagnesium bromide reacts with methyl p-chlorobenzoate to give p-nonylbenzoic acid, in the presence of Tris(acetylacetonato)iron(III) (Fe(acac)3), after workup with NaOH to hydrolyze the ester, shown as follows. Without the Fe(acac)3, the Grignard reagent would attack the ester group over the aryl halide.[26]

For the coupling of aryl halides with aryl Grignard reagents, nickel chloride in tetrahydrofuran (THF) is also a good catalyst. Additionally, an effective catalyst for the couplings of alkyl halides is dilithium tetrachlorocuprate (Li2CuCl4), prepared by mixing lithium chloride (LiCl) and copper(II) chloride (CuCl2) in THF. The Kumada-Corriu coupling gives access to [substituted] styrenes.
Oxidation
Treatment of a Grignard reagent with oxygen gives the magnesium organoperoxide. Hydrolysis of this material yields hydroperoxides or alcohol. These reactions involve radical intermediates.

The simple oxidation of Grignard reagents to give alcohols is of little practical import as yields are generally poor. In contrast, two-step sequence via a borane (vide supra) that is subsequently oxidized to the alcohol with hydrogen peroxide is of synthetic utility.
The synthetic utility of Grignard oxidations can be increased by a reaction of Grignard reagents with oxygen in presence of an alkene to an ethylene extended alcohol.[27] This modification requires aryl or vinyl Grignards. Adding just the Grignard and the alkene does not result in a reaction demonstrating that the presence of oxygen is essential. The only drawback is the requirement of at least two equivalents of Grignard although this can partly be circumvented by the use of a dual Grignard system with a cheap reducing Grignard such as n-butylmagnesium bromide.

Elimination
In the Boord olefin synthesis, the addition of magnesium to certain β-haloethers results in an elimination reaction to the alkene. This reaction can limit the utility of Grignard reactions.

Degradation of Grignard reagents
At one time, the formation and hydrolysis of Grignard reagents was used in the determination of the number of halogen atoms in an organic compound.[28] In modern usage Grignard degradation is used in the chemical analysis of certain triacylglycerols.[29]
Industrial use
An example of the Grignard reaction is a key step in the (non-stereospecific) industrial production of Tamoxifen[30] (currently used for the treatment of estrogen receptor positive breast cancer in women):[31]

Gallery
 Magnesium turnings are placed in a flask.
Magnesium turnings are placed in a flask. Tetrahydrofuran and a small piece of iodine are added.
Tetrahydrofuran and a small piece of iodine are added. A solution of alkyl bromide is added while heating.
A solution of alkyl bromide is added while heating. After completion of the addition, the mixture is heated for a while.
After completion of the addition, the mixture is heated for a while. Formation of the Grignard reagent is complete. A small amount of magnesium still remains in the flask.
Formation of the Grignard reagent is complete. A small amount of magnesium still remains in the flask. The Grignard reagent thus prepared is cooled to 0°C before the addition of the carbonyl compound. The solution becomes cloudy as the Grignard reagent precipitates out.
The Grignard reagent thus prepared is cooled to 0°C before the addition of the carbonyl compound. The solution becomes cloudy as the Grignard reagent precipitates out.- A solution of carbonyl compound is added to the Grignard reagent.
 The solution is warmed to room temperature. At this point the reaction is complete.
The solution is warmed to room temperature. At this point the reaction is complete.
See also
| Wikimedia Commons has media related to Grignard reactions. |
- Wittig reaction
- Barbier reaction
- Bodroux-Chichibabin aldehyde synthesis
- Fujimoto-Belleau reaction
- Organolithium reagents
- Sakurai reaction
- Indium mediated allylation
- Alkynylation
References
- ↑ Smith, Michael B.; March, Jerry (2007), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure (6th ed.), New York: Wiley-Interscience, ISBN 0-471-72091-7
- ↑ Chapter 19: Carboxylic Acids. Organic Chemistry 4e Carey. mhhe.com
- ↑ Shirley, D. A. (1954). "The Synthesis of Ketones from Acid Halides and Organometallic Compounds of Magnesium, Zinc, and Cadmium". Org. React. 8: 28–58.
- ↑ Huryn, D. M. (1991). "Carbanions of Alkali and Alkaline Earth Cations: (ii) Selectivity of Carbonyl Addition Reactions". In Trost, B. M.; Fleming, I. Comprehensive Organic Synthesis, Volume 1: Additions to C—X π-Bonds, Part 1. Elsevier Science. pp. 49–75. ISBN 978-0-08-052349-1. doi:10.1016/B978-0-08-052349-1.00002-0.
- ↑ IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). ISBN 0-9678550-9-8. doi:10.1351/goldbook.
- ↑ Grignard, V. (1900). "Sur quelques nouvelles combinaisons organométaliques du magnésium et leur application à des synthèses d'alcools et d'hydrocabures". Compt. Rend. 130: 1322–25.
- ↑ Maruyama, K.; Katagiri, T. (1989). "Mechanism of the Grignard reaction". J. Phys. Org. Chem. 2 (3): 205–213. doi:10.1002/poc.610020303.
- ↑ Garst, J. F.; Ungvary, F. "Mechanism of Grignard reagent formation". In Grignard Reagents; Richey, R. S., Ed.; John Wiley & Sons: New York, 2000; pp 185–275. ISBN 0-471-99908-3.
- ↑ Advanced Organic chemistry Part B: Reactions and Synthesis F.A. Carey, R.J. Sundberg 2nd Ed. 1983
- ↑ Rogers, H. R.; Hill, C. L.; Fujiwara, Y.; Rogers, R. J.; Mitchell, H. L.; Whitesides, G. M. (1980). "Mechanism of formation of Grignard reagents. Kinetics of reaction of alkyl halides in diethyl ether with magnesium". Journal of the American Chemical Society. 102 (1): 217. doi:10.1021/ja00521a034.
- ↑ De Boer, H.J.R.; Akkerman, O.S; Bickelhaupt, F. (1988). "Carbanions as intermediates in the synthesis of Grignard Reagents". Angew. Chem. Int. Ed. 27 (5): 687–89. doi:10.1002/anie.198806871.
- ↑ Van Klink, G.P.M.; de Boer, H.J.R; Schat, G.; Akkerman, O.S.; Bickelhaupt, F.; Spek, A. (2002). "Carbanions as Intermediates in the Formation of Grignard Reagents". Organometallics. 21 (10): 2119–35. doi:10.1021/om011083a.
- ↑ Silverman, G. S., Rakita, P. E. (eds.) (1996) Handbook of Grignard Reagents. CRC Press. ISBN 0824795458
- ↑ Goebel, M. T.; Marvel, C. S. (1933). "The Oxidation of Grignard Reagents". Journal of the American Chemical Society. 55 (4): 1693–1696. doi:10.1021/ja01331a065.
- ↑ Smith, David H. (1999). "Grignard Reactions in "Wet" Ether". Journal of Chemical Education. 76 (10): 1427. Bibcode:1999JChEd..76.1427S. doi:10.1021/ed076p1427.
- ↑ Lai Yee Hing (1981). "Grignard Reagents from Chemically Activated Magnesium". Synthesis. 1981 (8): 585–604. doi:10.1055/s-1981-29537.
- ↑ Clayden, Jonathan; Greeves, Nick (2005). Organic chemistry. Oxford: Oxford Univ. Press. p. 212. ISBN 978-0-19-850346-0.
- ↑ Krasovskiy, Arkady; Knochel, Paul. "Convenient Titration Method for Organometallic Zinc, Magnesium, and Lanthanide Reagents". Synthesis. 2006: 890–891. doi:10.1055/s-2006-926345.
- ↑ Smith, David H. (1999). "Grignard Reactions in "Wet" Ether". Journal of Chemical Education. 76 (10): 1427. Bibcode:1999JChEd..76.1427S. doi:10.1021/ed076p1427.
- ↑ Philip E. Rakita (1996). "5. Safe Handling Practices of Industrial Scale Grignard Ragents". In Gary S. Silverman; Philip E. Rakita. Handbook of Grignard reagents (Google Books excerpt). CRC Press. pp. 79–88. ISBN 0-8247-9545-8.
- ↑ Knochel, P.; Dohle, W.; Gommermann, N.; Kneisel, F. F.; Kopp, F.; Korn, T.; Sapountzis, I.; Vu, V. A. (2003). "Highly Functionalized Organomagnesium Reagents Prepared through Halogen–Metal Exchange". Angewandte Chemie International Edition. 42: 4302–4320. PMID 14502700. doi:10.1002/anie.200300579.
- ↑ Henry Gilman and R. H. Kirby (1941). "Butyric acid, α-methyl-". Org. Synth.; Coll. Vol., 1, p. 361
- ↑ Haugan, Jarle André; Songe, Pål; Rømming, Christian; Rise, Frode; Hartshorn, Michael P.; Merchán, Manuela; Robinson, Ward T.; Roos, Björn O.; Vallance, Claire; Wood, Bryan R. (1997). "Total Synthesis of C31-Methyl Ketone Apocarotenoids 2: The First Total Synthesis of (3R)-Triophaxanthin" (PDF). Acta Chemica Scandinavica. 51: 1096–1103. doi:10.3891/acta.chem.scand.51-1096. Retrieved 2009-11-26.
- ↑ Peters, D. G.; Ji, C. (2006). "A Multistep Synthesis for an Advanced Undergraduate Organic Chemistry Laboratory". Journal of Chemical Education. 83 (2): 290. doi:10.1021/ed083p290.
- ↑ "Unit 12 Aldehydes, Ketones and Carboxylic Acids". Chemistry Part II Textbook for class XII (PDF). 2. India: National Council of Educational Research and Training. 2010. p. 355. ISBN 81-7450-716-7.
- ↑ A. Fürstner, A. Leitner, G. Seidel (2004). "4-Nonylbenzoic Acid". Org. Synth. 81: 33–42.
- ↑ Youhei Nobe; Kyohei Arayama; Hirokazu Urabe (2005). "Air-Assisted Addition of Grignard Reagents to Olefins. A Simple Protocol for a Three-Component Coupling Process Yielding Alcohols". J. Am. Chem. Soc. 127 (51): 18006–18007. PMID 16366543. doi:10.1021/ja055732b.
- ↑ Steinkopf, Wilhelm; V. Petersdorff, Hans-JüRgen (1940). "Studien in der Thiophenreihe. LI. Atophanartige Derivate des Dithienyls und Diphenyls". Justus Liebigs Ann. Chem. 543: 119–128. doi:10.1002/jlac.19405430110.
- ↑ Myher JJ; Kuksis A (February 1979). "Stereospecific analysis of triacylglycerols via racemic phosphatidylcholines and phospholipase C". Can. J. Biochem. 57 (2): 117–24. PMID 455112. doi:10.1139/o79-015.
- ↑ Richey, Herman Glenn (2000). Grignard Reagents: New Developments. Wiley. ISBN 0471999083.
- ↑ Jordan VC (1993). "Fourteenth Gaddum Memorial Lecture. A current view of tamoxifen for the treatment and prevention of breast cancer". Br J Pharmacol. 110 (2): 507–17. PMC 2175926
 . PMID 8242225. doi:10.1111/j.1476-5381.1993.tb13840.x.
. PMID 8242225. doi:10.1111/j.1476-5381.1993.tb13840.x.
Further reading
- ed. by Gary S. Silverman .... (1996). Rakita, Philip E.; Silverman, Gary, eds. Handbook of Grignard reagents. New York, N.Y: Marcel Dekker. ISBN 0-8247-9545-8.
- Mary McHale, "Grignard Reaction," Connexions, http://cnx.org/content/m15245/1.2/. 2007.
- Grignard knowledge: Alkyl coupling chemistry with inexpensive transition metals by Larry J. Westrum, Fine Chemistry November/December 2002, pp. 10–13