Protein superfamily
A protein superfamily is the largest grouping (clade) of proteins for which common ancestry can be inferred (see homology). Usually this common ancestry is inferred from structural alignment[1] and mechanistic similarity, even if no sequence similarity is evident.[2] Sequence homology can then be deduced even if not apparent (due to low sequence similarity). Superfamilies typically contain several protein families which show sequence similarity within each family. The term protein clan is commonly used for protease superfamilies based on the MEROPS protease classification system.[2]
Identification

Superfamilies of proteins are identified using a number of methods. Closely related members can be identified by different methods to those needed to group the most evolutionarily divergent members.
Sequence similarity

Historically, the similarity of different amino acid sequences has been the most common method of inferring homology.[4] Sequence similarity is considered a good predictor of relatedness, since similar sequences are more likely the result of gene duplication and divergent evolution, rather than the result of convergent evolution. Amino acid sequence is typically more conserved than DNA sequence (due to the degenerate genetic code), so is a more sensitive detection method. Since some of the amino acids have similar properties (e.g., charge, hydrophobicity, size), conservative mutations that interchange them are often neutral to function. The most conserved sequence regions of a protein often correspond to functionally important regions like catalytic sites and binding sites, since these regions are less tolerant to sequence changes.
Using sequence similarity to infer homology has several limitations. There is no minimum level of sequence similarity guaranteed to produce identical structures. Over long periods of evolution, related proteins may show no detectable sequence similarity to one another. Sequences with many insertions and deletions can also sometimes be difficult to align and so identify the homologous sequence regions. In the PA clan of proteases, for example, not a single residue is conserved through the superfamily, not even those in the catalytic triad. Conversely, the individual families that make up a superfamily are defined on the basis of their sequence alignment, for example the C04 protease family within the PA clan.
Nevertheless, sequence similarity is the most commonly used form of evidence to infer relatedness, since the number of known sequences vastly outnumbers the number of known tertiary structures.[5] In the absence of structural information, sequence similarity constrains the limits of which proteins can be assigned to a superfamily.[5]
Structural similarity
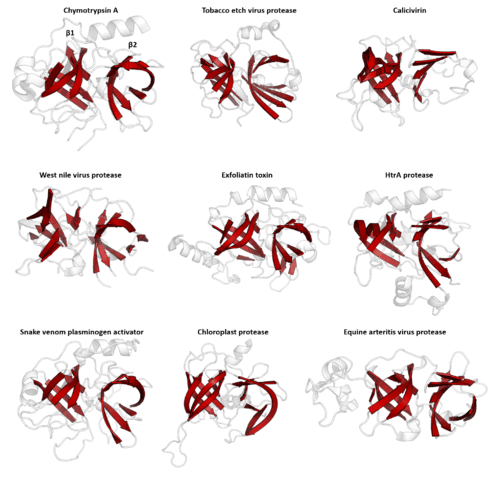
Structure is much more evolutionarily conserved than sequence, such that proteins with highly similar structures can have entirely different sequences. Over very long evolutionary timescales, very few residues show detectable amino acid sequence conservation, however secondary structural elements and tertiary structural motifs are highly conserved. Conformational changes of the protein structure may also be conserved, as is seen in the serpin superfamily. Consequently, protein tertiary structure can be used to detect homology between proteins even when no evidence of relatedness remains in their sequences. Structural alignment programs, such as DALI, use the 3D structure of a protein of interest to find proteins with similar folds. However, on rare occasions, related proteins may evolve to be structurally dissimilar and relatedness can only be inferred by other methods.
Mechanistic similarity
The catalytic mechanism of enzymes within a superfamily is typically conserved, although substrate specificity may be significantly different. Catalytic residues also tend to occur in the same order in the protein sequence. The families within the PA clan of proteases, although there has been divergent evolution of the catalytic triad residues used to perform catalysis, all members use a similar mechanism to perform covalent, nucleophilic catalysis on proteins, peptides or amino acids. However, mechanism alone is not sufficient to infer relatedness, since some catalytic mechanisms have been convergently evolved multiple times independently, and so form separate superfamilies.[6]
Evolutionary significance
Protein superfamilies represent the current limits of our ability to identify common ancestry.[7] They are the largest evolutionary grouping based on direct evidence that is currently possible. They are therefore amongst the most ancient evolutionary events currently studied. Some superfamilies have members present in all kingdoms of life, indicating that the last common ancestor of that superfamily was in the last universal common ancestor of all life (LUCA).[8]
Superfamily members may be in different species, with the ancestral protein being the form of the protein that existed in the ancestral species (orthology). Conversely, the proteins may be in the same species, but evolved from a single protein whose gene was duplicated in the genome (paralogy).
Diversification
A majority of proteins contain multiple domains. Between 66-80% of eukaryotic proteins have multiple domains while about 40-60% of prokaryotic proteins have multiple domains.[4] Over time, many of the superfamilies of domains have mixed together. In fact, it is very rare to find “consistently isolated superfamilies”.[4] When domains do combine, the N- to C- terminal domain order (the "domain architecture") is typically well conserved. Additionally, the number of domain combinations seen in nature is small compared to the number of possibilities, suggesting that selection acts on all combinations.[4]
Examples
α/β hydrolase superfamily - Members share an α/β sheet, containing 8 strands connected by helices, with catalytic triad residues in the same order,[9] activities include proteases, lipases, peroxidases, esterases, epoxide hydrolases and dehalogenases.[10]
Alkaline phosphatase superfamily - Members share an αβα sandwich structure[11] as well as performing common promiscuous reactions by a common mechanism.[12]
Globin superfamily - Members share an 8-alpha helix globular globin fold.[13][14]
Immunoglobulin superfamily - Members share a sandwich-like structure of two sheets of antiparallel β strands (Ig-fold), and are involved in recognition, binding, and adhesion.[15][16]
PA clan - Members share a chymotrypsin-like double β-barrel fold and similar proteolysis mechanisms but sequence identity of <10%. The clan contains both cysteine and serine proteases (different nucleophiles).[2][17]
Ras superfamily - Members share a common catalytic G domain of a 6-strand β sheet surrounded by 5 α-helices.[18]
Serpin superfamily - Members share a high-energy, stressed fold which can undergo a large conformational change, which is typically used to inhibit serine and cysteine proteases by disrupting their structure.[19]
TIM barrel superfamily - Members share a large α8β8 barrel structure. It is one of the most common protein folds and the monophylicity of this superfamily is still contested.[20][21]
Protein superfamily resources
Several biological databases document protein superfamilies and protein folds, for example:
- Pfam - Protein families database of alignments and HMMs
- PROSITE - Database of protein domains, families and functional sites
- PIRSF - SuperFamily Classification System
- PASS2 - Protein Alignment as Structural Superfamilies v2
- SUPERFAMILY - Library of HMMs representing superfamilies and database of (superfamily and family) annotations for all completely sequenced organisms
- SCOP and CATH - Classifications of protein structures into superfamilies, families and domains
Similarly there are algorithms that search the PDB for proteins with structural homology to a target structure, for example:
- DALI - Structural alignment based on a distance alignment matrix method
See also
- Structural alignment
- Protein domains
- Protein family
- Protein mimetic
- Protein structure
- Homology (biology)
- Interolog
- List of gene families
- SUPERFAMILY
- CATH
References
- ↑ Holm, L; Rosenström, P (July 2010). "Dali server: conservation mapping in 3D.". Nucleic Acids Research. 38 (Web Server issue): W545–9. PMC 2896194
 . PMID 20457744. doi:10.1093/nar/gkq366.
. PMID 20457744. doi:10.1093/nar/gkq366. - 1 2 3 Rawlings, ND; Barrett, AJ; Bateman, A (January 2012). "MEROPS: the database of proteolytic enzymes, their substrates and inhibitors.". Nucleic Acids Research. 40 (Database issue): D343–50. PMC 3245014 . PMID 22086950. doi:10.1093/nar/gkr987.
- ↑ "Clustal FAQ #Symbols". Clustal. Retrieved 8 December 2014.
- 1 2 3 4 Han, JH (2007). "The folding and evolution of multidomain proteins.". Nature Reviews Molecular Cell Biology. 8 (4): 319–30. PMID 17356578. doi:10.1038/nrm2144.
- 1 2 Pandit, SB, Gosar D, Abhiman S; et al. (2002). "SUPFAM--a database of potential protein superfamily relationships derived by comparing sequence-based and structure-based families: implications for structural genomics and function annotation in genomes". Nucleic Acids Research. 30 (1): 289–293. doi:10.1093/nar/30.1.289.
- ↑ Buller, Andrew R.; Townsend, Craig A. (2013-02-19). "Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad". Proceedings of the National Academy of Sciences. 110 (8): E653–E661. ISSN 0027-8424. PMC 3581919 . PMID 23382230. doi:10.1073/pnas.1221050110.
- ↑ Shakhnovich, BE; Deeds, E; Delisi, C; Shakhnovich, E (March 2005). "Protein structure and evolutionary history determine sequence space topology.". Genome Research. 15 (3): 385–92. PMC 551565 . PMID 15741509. doi:10.1101/gr.3133605.
- ↑ Ranea, JA; Sillero, A; Thornton, JM; Orengo, CA (October 2006). "Protein superfamily evolution and the last universal common ancestor (LUCA).". Journal of Molecular Evolution. 63 (4): 513–25. PMID 17021929. doi:10.1007/s00239-005-0289-7.
- ↑ Carr PD, Ollis DL (2009). "α/β hydrolase fold: an update". Protein Pept. Lett. 16 (10): 1137–48. PMID 19508187.
- ↑ Nardini M, Dijkstra BW (December 1999). "α/β hydrolase fold enzymes: the family keeps growing". Curr. Opin. Struct. Biol. 9 (6): 732–7. PMID 10607665. doi:10.1016/S0959-440X(99)00037-8.
- ↑ "SCOP". Retrieved 28 May 2014.
- ↑ Mohamed, MF; Hollfelder, F (Jan 2013). "Efficient, crosswise catalytic promiscuity among enzymes that catalyze phosphoryl transfer.". Biochimica et Biophysica Acta. 1834 (1): 417–24. PMID 22885024. doi:10.1016/j.bbapap.2012.07.015.
- ↑ Branden, Carl; Tooze, John (1999). Introduction to protein structure (2nd ed.). New York: Garland Pub. ISBN 978-0815323051.
- ↑ Bolognesi, M; Onesti, S; Gatti, G; Coda, A; Ascenzi, P; Brunori, M (1989). "Aplysia limacina myoglobin. Crystallographic analysis at 1.6 a resolution". Journal of Molecular Biology. 205 (3): 529–44. PMID 2926816. doi:10.1016/0022-2836(89)90224-6.
- ↑ Bork P, Holm L, Sander C (September 1994). "The immunoglobulin fold. Structural classification, sequence patterns and common core". J. Mol. Biol. 242 (4): 309–20. PMID 7932691. doi:10.1006/jmbi.1994.1582.
- ↑ Brümmendorf T, Rathjen FG (1995). "Cell adhesion molecules 1: immunoglobulin superfamily". Protein Profile. 2 (9): 963–1108. PMID 8574878.
- ↑ Bazan, JF; Fletterick, RJ (November 1988). "Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications.". Proceedings of the National Academy of Sciences of the United States of America. 85 (21): 7872–6. PMC 282299 . PMID 3186696. doi:10.1073/pnas.85.21.7872.
- ↑ Vetter IR, Wittinghofer A (November 2001). "The guanine nucleotide-binding switch in three dimensions". Science. 294 (5545): 1299–304. PMID 11701921. doi:10.1126/science.1062023.
- ↑ Silverman, G. A.; Bird, P. I.; Carrell, R. W.; Church, F. C.; Coughlin, P. B.; Gettins, P. G. W.; Irving, J. A.; Lomas, D. A.; Luke, C. J.; Moyer, R. W.; Pemberton, P. A.; Remold-O'Donnell, E.; Salvesen, G. S.; Travis, J.; Whisstock, J. C. (2 July 2001). "The Serpins Are an Expanding Superfamily of Structurally Similar but Functionally Diverse Proteins: EVOLUTION, MECHANISM OF INHIBITION, NOVEL FUNCTIONS, AND A REVISED NOMENCLATURE". Journal of Biological Chemistry. 276 (36): 33293–33296. PMID 11435447. doi:10.1074/jbc.R100016200.
- ↑ Nagano, N; Orengo, CA; Thornton, JM (Aug 30, 2002). "One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions.". Journal of Molecular Biology. 321 (5): 741–65. PMID 12206759. doi:10.1016/s0022-2836(02)00649-6.
- ↑ Farber, G (1993). "An α/β-barrel full of evolutionary trouble". Current Opinion in Structural Biology. 3 (3): 409–412. doi:10.1016/S0959-440X(05)80114-9.