Enantioselective reduction of ketones
Enantioselective ketone reductions convert prochiral ketones into chiral, non-racemic alcohols and are used heavily for the synthesis of stereodefined alcohols.[1]
Introduction
Carbonyl reduction, the net addition of H2 across a carbon-oxygen double bond, is a straightforward way to generate alcohols. Stoichiometric reducing agents to accomplish this task include lithium aluminium hydride, sodium borohydride, alkoxy borohydrides, alkoxy aluminium hydrides, and boranes. Initial efforts toward enantioselective ketone reductions focused on the development of chiral, non-racemic reducing agents. Although stoichiometric chiral reducing agents often afford products with high enantioselectivity, the necessity of a stoichiometric amount of chiral material is a disadvantage of these reagents.[2]
The catalytic, asymmetric reduction of ketones may be accomplished through the use of catalytic amounts of an oxazaborolidine catalyst in conjunction with borane or catecholborane as the stoichiometric reducing agent.[3] Oxazaborolidines remain in common use for reductions of simple ketones.
More recently, efforts in the field of enantioselective reduction have focused on the development of transition metal catalyzed reactions, which employ cheap reductants such as hydrogen gas (H2), formic acid (HCO2H), or isopropanol ((CH3)2CHOH). The latter two reagents are used for transfer hydrogenations, which represent the formal transfer of an H2 molecule from the reductant to the substrate.[4] Asymmetric induction in transition metal catalyzed reactions is achieved through the use of a chiral Lewis basic ligand in catalytic amounts. For ketone substrates that can chelate the metal catalyst, enantioselectivities of transition metal catalyzed reactions may be higher (and side reactions less prevalent) than the corresponding oxazaborolidine reductions.[5]
(1)

Mechanism and Stereochemistry
Oxazaborolidine Reductions
The mechanism of oxazaborolidine reductions has been supported by ab initio calculations.[6] Coordination of borane to the oxazaborolidine nitrogen generates the complex I, which then coordinates a molecule of ketone to yield complex II. In the transition state for hydride transfer (II → III), the large substituent of the ketone is aligned inward to avoid steric interactions with the outward-pointing R group of the oxazaborolidine, which is often tethered to the nitrogen atom. After hydride transfer, complex III releases the product and coordinates a second molecule of borane.
(2)

Transition Metal Catalyzed Reductions
Transition metal catalyzed reductions may proceed by a variety of mechanisms, depending on the reductant and metal employed. Regardless of the precise mechanism, it is the spatial properties of the chiral ligand bound to the metal center that determine the sense and extent of enantioselectivity. A reliable stereochemical model has been developed for reductions employing BINAP ligands.[7] When BINAP chelates to a transition metal such as ruthenium, the phenyl groups attached to phosphorus reside in either pseudoaxial or psudeoequatorial positions. The pseudoequatorial phenyl groups project into the region of space on the other side of the BINAP ligand and influence the preferred binding conformation of chelating ketones (such as α-amino ketones or β-keto esters). The ketone typically occupies the more open regions of space, leading to hydride delivery to a single face of the ketone. The C2 symmetry of the coordination space ensures that only a single face of the ketone will be accessible to the catalyst, no matter in which open region the ketone binds.
(3)

Scope and Limitations
Stoichiometric, Chiral Hydride Reductions
Lithium aluminium hydride (LAH) modified with chiral alkoxide ligands may be used to synthesize chiral alcohols in good yield and high enantioselectivity. Chelating ligands such as BINOL[8] are used to avoid disproportionation and background reduction by LAH. Chiral diamines and amino alcohols have also been used to modify LAH for enantioselective reductions.
(4)

Chirally modified borohydrides are also useful for enantioselective ketone reductions. Economical ligands derived from amino acids have been used to modify borohydrides, affording highly selective reducing agents.[9]
(5)

Chiral alkylborohydrides are accessible through the diastereoselective hydroboration of chiral alkenes. Boranes derived from pinene have been used in this context for enantioselective reductions.[10] Neutral alkoxyboranes may result from these reductions.
(6)

Catalytic Reductions of Ketones
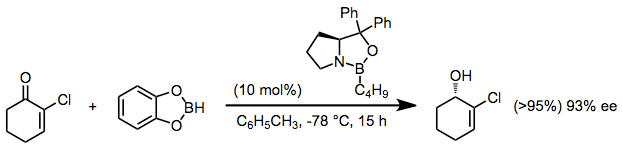
With borane or catecholborane as the stoichiometric reducing agent, chiral oxazaborolidine catalysts may be used to reduce ketones enantioselectively. Catecholborane may be used as an alternative to solutions of borane-Lewis base adducts.[11]
(7)
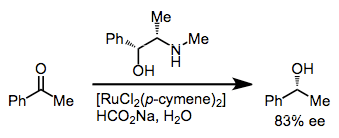
Reduction via the net transfer of hydrogen from one organic molecule to another is known as transfer hydrogenation. Transfer hydrogenation to ketones leads to alcohols (the Meerwein-Ponndorf-Verley reduction), and in the presence of a chiral transition metal catalyst, this process may be rendered enantioselective. In the presence of a chiral diamine, ruthenium catalyzes the enantioselective transfer hydrogenation of aryl ketones with isopropanol.[12] Other metals that have been employed include samarium(III),[13] iridium(I),[14] and rhodium(I).[15]
(8)

Formic acid and formate salts may also be used as reductants in transfer hydrogenations. Simple aryl ketones are reduced enantioselectively when a chiral amino alcohol ligand is employed.[4]
(9)
Transition metal catalysts have also been used with hydrogen gas as the stoichiometric reductant. Ketones with a chelating group undergo enantioselective reduction in the presence of a chiral Ru(BINAP) catalyst.[16] The configuration of the new stereocenter is predictable using the stereochemical model developed for hydrogenations employing BINAP (see equation (3) above).
(10)

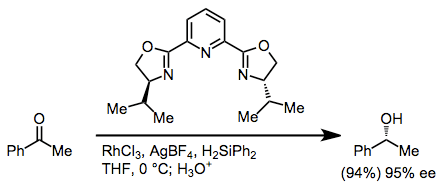
Hydrosilylation may be used to reduce ketones after silyl ether hydrolysis. Rhodium(I) and rhodium(III) salts are the most common catalysts for hydrosilylation. Asymmetric induction may be conferred by chiral PYBOX ligands.[17]
(11)
Enzymatic Reductions
Microorganisms reduce certain classes of simple ketones with extremely high enantioselectivity. Baker's yeast is the most common organism used to reduce ketones enzymatically,[18][19] although other microorganisms may be used. Access to "unnatural" reduced enantiomers is difficult in most cases.
(12)

Experimental Conditions and Procedure
Example Procedure[20]
(13)

(S,S)-1,2-Diphenylethylenediamine (122) (7.5 mg, 0.035 mmol) and a 0.5 M 2-propanol solution of KOH (140 μL, 0.070 mmol) were added to 2-propanol (10 mL) and the mixture was degassed by freeze-thaw cycles. To this solution was added RuCl2[(S)-BINAP](dmf)n (269) (33.1 mg, 0.035 mmol), and the resulting mixture was sonicated for 10 minutes and used as a catalyst. A solution of 1-acetonaphthone (30.0 g, 176 mmol) in 2-propanol (90 mL) was subjected to freeze-thaw cycles. These two solutions were transferred to a glass autoclave, hydrogen was pressurized to 8 atm, and the solution was vigorously stirred at 28° for 24 hours. After venting hydrogen, the solvent was removed under reduced pressure, and the residue was distilled to give (R)-1-(1-naphthyl)ethanol (27.90 g, 92% yield, 95% ee), bp 98–100°/0.5 mmHg, [α]25D + 75.8° (c 0.99, ether) (lit. (270) [α]25D + 82.1° (c 1.0, ether)). The purity determined by 1H NMR was > 99%. 1H NMR (CDCl3/TMS): δ 1.64 (d, J = 6 Hz, 3 H), 1.95 (bs, 1 H), 5.64 (q, J = 6 Hz, 1 H), 7.43–8.10 (m, 7 H); 13C NMR (CDCl3/TMS): δ 25.50, 70.56, 123.9, 124.1, 126.5, 126.8, 128.2,128.9, 132.6, 134.0, 134.4, 142.8.[21]
References
- ↑ Itsuno, S. Org. React. 1998, 52, 395. doi:10.1002/0471264180.or052.02
- ↑ Yamamoto, K.; Fukushima, H.; Nakazaki, M. J. Chem. Soc., Chem. Commun. 1984, 1490.
- ↑ Hirao, A.; Itsuno, S.; Nakahama, S.; Yamazaki, N. J. Chem. Soc., Chem. Commun. 1981, 315.
- 1 2 Mao, J.; Wan, B.; Wu, F.; Lu, S. Tetrahedron Lett. 2005, 46, 7341.
- ↑ Giacomelli, G.; Lardicci, L.; Palla, F. J. Org. Chem. 1984, 49, 310.
- ↑ Nevalainen, V. Tetrahedron: Asymmetry 1992, 3, 1441.
- ↑ Imamoto, T. Chem. Commun., 2009, 7447.
- ↑ Chan, P. C.-M.; Chong, J. M. J. Org. Chem. 1988, 53, 5586.
- ↑ Soai, K. J. Synth. Org. Chem., Jpn. 1989, 47, 11.
- ↑ Ramachandran, P. V.; Brown, H. C.; Swaminathan, S. Tetrahedron: Asymmetry 1990, 1, 433.
- ↑ Corey, E. J.; Balzski, R. K. Tetrahedron Lett. 1990, 31, 611.
- ↑ Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 7562.
- ↑ Evans, D. A.; Nelson, S. G.; Gagné, M. R.; Muci, A. R. J. Am. Chem. Soc. 1993, 115, 9800.
- ↑ Zassinovich, G.; Bettella, R.; Mestroni, G.; Brestiani-Pahor, N.; Geremia, S.; Randaccio, L. J. Organomet. Chem. 1989, 370, 187.
- ↑ Gladiali, S.; Pinna, L.; Delogu, G.; De Martin, S.; Zassinovich, G.; Mestroni, G. Tetrahedron: Asymmetry 1990, 1, 635.
- ↑ Kitamura, M.; Okuma, T.; Inoue, S.; Sayo, N.; Kumobayashi, H.; Akutagawa, S.; Ohta, T.; Takaya, H.; Noyori, R. J. Am. Chem. Soc. 1988, 110, 629.
- ↑ Nishiyama, H.; Kondo, M. Nakamura, T.; Itoh, K. Organometallics 1991, 10, 500.
- ↑ Csuk, Rene.; Glaenzer, Brigitte I. (1991-01-01). "Baker's yeast mediated transformations in organic chemistry". Chemical Reviews. 91 (1): 49–97. ISSN 0009-2665. doi:10.1021/cr00001a004.
- ↑ Inoue, T.; Hosomi, K.; Araki, M.; Nishide, K.; Node, M. Tetrahedron: Asymmetry 1995, 6, 31 doi:10.1016/0957-4166(94)00344-B.
- ↑ Ohkuma, T.; Ooka, H.; Hashiguchi, S.; Ikariya, T.; Noyori, R. J. Am. Chem. Soc. 1995, 117, 2675.
- ↑ Li, X.; Wu, X.; Chen, W.; Hancock, F.; King, F.; Xiao, J. Org. Lett. 2004, 6, 3321.