Dioxygenase
| Dioxygenase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
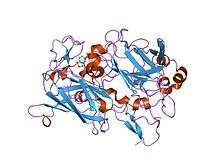 crystal structure of acinetobacter sp. adp1 protocatechuate 3,4-dioxygenase in complex with 3,4-dihydroxybenzoate | |||||||||
| Identifiers | |||||||||
| Symbol | Dioxygenase_C | ||||||||
| Pfam | PF00775 | ||||||||
| Pfam clan | CL0287 | ||||||||
| InterPro | IPR000627 | ||||||||
| PROSITE | PDOC00079 | ||||||||
| SCOP | 2pcd | ||||||||
| SUPERFAMILY | 2pcd | ||||||||
| |||||||||
Dioxygenases are oxidoreductase enzymes. Aerobic life, from simple single-celled bacteria species to complex eukaryotic organisms, has evolved to depend on the oxidizing power of dioxygen in various metabolic pathways. From energetic adenosine triphosphate (ATP) generation to xenobiotic degradation, the use of dioxygen as a biological oxidant is widespread and varied in the exact mechanism of its use. Enzymes employ many different schemes to use dioxygen, and this largely depends on the substrate and reaction at hand.
Comparison with monooxygenases
In the monooxygenases, only a single atom of dioxygen is incorporated into a substrate with the other being reduced to a water molecule. The dioxygenases (EC 1.13.11) catalyze the oxidation of a substrate without the reduction of one oxygen atom from dioxygen into a water molecule. However, this definition is ambiguous because it does not take into account how many substrates are involved in the reaction. The majority of dioxygenases fully incorporate dioxygen into a single substrate, and a variety of cofactor schemes are utilized to achieve this. For example, in the α-ketoglutarate-dependent enzymes, one atom of dioxygen is incorporated into two substrates, with one always being α-ketoglutarate, and this reaction is brought about by a mononuclear iron center.
Iron-containing enzymes
The most widely observed cofactor involved in dioxygenation reactions is iron, but the catalytic scheme employed by these iron-containing enzymes is highly diverse. Iron-containing dioxygenases can be subdivided into three classes on the basis of how iron is incorporated into the active site: those employing a mononuclear iron center, those containing a Rieske [2Fe-2S] cluster, and those utilizing a heme prosthetic group.
Mononuclear iron dioxygenases
The mononuclear iron dioxygenases, or non-heme iron-dependent dioxygenases as they are also termed, all utilize a single catalytic iron to incorporate either one or both atoms of dioxygen into a substrate. Despite this common oxygenation event, the mononuclear iron dioxygenases are diverse in how dioxygen activation is used to promote certain chemical reactions.[1] For instance, carbon-carbon bond cleavage, fatty acid hydroperoxidation, carbon-sulfur bond cleavage, and thiol oxidation are all reactions catalyzed by mononuclear iron dioxygenases.[1][2][3]
Most mononculear iron dioxygenases are members of the cupin superfamily in which the overall domain structure is described as a six-stranded β-barrel fold (or jelly roll motif). At the center this barrel structure is a metal ion, most commonly ferrous iron, whose coordination environment is frequently provided by residues in two partially conserved structural motifs: G(X)5HXH(X)3-4E(X)6G and G(X)5-7PXG(X)2H(X)3N.[4][5]
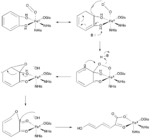
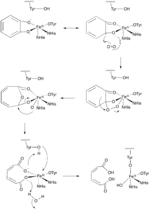
Two important groups of mononuclear, non-heme iron dioxygenases are catechol dioxygenases and 2-oxoglutarate (2OG)-dependent dioxygenases.[6] The catechol dioxygenases, some of the most well-studied dioxygenase enzymes, use dioxygen to cleave a carbon-carbon bond of an aromatic catechol ring system.[4] Catechol dioxygenases are further classified as being “extradiol” or “intradiol,” and this distinction is based on mechanistic differences in the reactions (figures 1 & 2). Intradiol enzymes cleave the carbon-carbon bond between the two hydroxyl groups. The active ferric center is coordinated by four protein ligands—two histidine and two tyrosinate residues—in a trigonal bipyramidal manner with a water molecule occupying the fifth coordination site.[3] Once a catecholate substrate binds to the metal center in a bidentate fashion through the deprotonated hydroxyl groups, the ferric iron “activates” the substrate by means of abstracting an electron to produce a radical on the substrate. This then allows for reaction with dioxygen and subsequent intradiol cleavage to occur through a cyclic anhydride intermediate.[2][4] Extradiol members utilize ferrous iron as the active redox state, and this center is commonly coordinated octahedrally through a 2-His-1-Glu motif with labile water ligands occupying empty positions. Once a substrate binds to the ferrous center, this promotes dioxygen binding and subsequent activation.[2][4][7] This activated oxygen species then proceeds to react with the substrate ultimately cleaving the carbon-carbon bond adjacent to the hydroxyl groups through the formation of an α-keto lactone intermediate.[3]
In the 2OG-dependent dioxygenases, ferrous iron (Fe(II)) is also coordinated by a (His)2(Glu/Asp)1 "facial triad" motif. Bidentate coordination of 2OG and water completes a pseudo-octahedral coordination sphere. Following substrate binding, the water ligand is released, yielding an open coordination site for oxygen activation.[6] Upon oxygen binding, a poorly understood transformation occurs during which 2OG is oxidatively decarboxylated to succinate and the O-O bond is cleaved to form a Fe(IV)-oxo (ferryl) intermediate. This powerful oxidant is then utilized to carry out various reactions, including hydroxylation, halogenation, and demethylation.[8] In the best characterized case, the hydroxylases, the ferryl intermediate abstracts a hydrogen atom from the target position of the substrate, yielding a substrate radical and Fe(III)-OH. This radical then couples to the hydroxide ligand, producing the hydroxylated product and the Fe(II) resting state of the enzyme.[8]
Rieske dioxygenases
The Rieske dioxygenases catalyze the cis-dihydroxylation of arenes to cis-dihydro-diol products. These enzymes are prominently found in soil bacteria such as Pseudomonas,[3] and their reactions constitute the initial step in aromatic hydrocarbon biodegradation.[2] Rieske dioxygenases are structurally more complex than other dioxygenases due to the need for an efficient electron transfer pathway (figure 2) to mediate the additional, simultaneous two-electron reduction of the aromatic substrate.

A catalytically-competent Rieske dioxygenase has three components: an NADH-dependent FAD reductase, a ferredoxin with two [2Fe-2S] Rieske clusters, and an α3β3 oxygenase with each α-subunit containing a mononuclear iron center and a [2Fe-2S] Rieske cluster.[2] Within each α-subunit, the iron-sulfur cluster and mononuclear iron center are separated by a distance of some ~43 Å, much too far for efficient electron transfer to occur. Instead, it is proposed electron transfer is mediated through these two centers in adjacent subunits, that the iron-sulfur cluster of one subunit transfers electrons to the mononuclear iron center of the adjacent subunit which is conveniently separated by ~12 Å. While this distance would appear optimal for efficient electron transfer, replacement of the bridging aspartate residue causes a loss of enzyme function, suggesting that electron transfer instead proceeds through the hydrogen-bonding network held in place by this aspartate residue.[3]
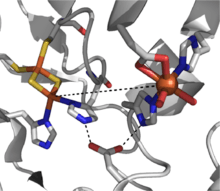
The mechanistic picture for this class of dioxygenases is not yet clear, but there is evidence supporting an iron(III) hydroperoxy intermediate in the reaction pathway.[7] This species could represent the active oxidant, or it could undergo hemolytic O-O bond cleavage to yield an iron(V)-oxo intermediate as the working oxidizing agent.[3][7] The Rieske dioxygenase are a powerful class of redox-active enzymes, and reactions such as sulfoxidation, desaturation, and benzylic oxidation have been reported in addition to dioxygenation.[2]
Heme-containing dioxygenases
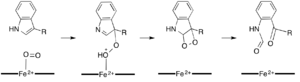
While most iron-dependent dioxygenases utilize a non-heme iron cofactor, the oxidation of L-(and D-)tryptophan to N-formylkynurenine is catalyzed by either tryptophan 2,3-dioxygenase (TDO) or indoleamine 2,3-dioxygenase (IDO), which are heme dioxygenases that utilize iron coordinated by a heme B prosthetic group.[9][10] While these dioxygenases are of interest in part because they uniquely use heme for catalysis, they are also of interest due to their importance in tryptophan regulation in the cell, which has numerous physiological implications.[11] The initial association of the substrate with the dioxygen-iron in the enzyme active site is thought to either proceed via radical or electrophilic addition, requiring either ferrous iron or ferric iron, respectively.[9] While the exact reaction mechanism for the heme-dependent dioxygenases is still under debate, it is postulated that the reaction proceeds through either a dioxetane or Criegee mechanism (figures 4, 5).[9][11]

Cambialistic dioxygenases
While iron is by far the most prevalent cofactor used for enzymatic dioxygenation, it is not required by all dioxygenases for catalysis. Quercetin 2,3-dioxygenase (quercetinase, QueD) catalyzes the dioxygenolytic cleavage of quercetin to 2-protocatechuoylphloroglucinolcarboxylic acid and carbon monoxide.[12] The most characterized enzyme, from Aspergillus japonicus, requires the presence of copper,[4] and bacterial quercetinases have been discovered that are quite promiscuous (cambialistic)[13] in their requirements of a metal center, with varying degrees of activity reported with substitution of divalent manganese, cobalt, iron, nickel and copper.[12] (Quercetin, role in metabolism). Acireductone (1,2-dihydroxy-5-(methylthio)pent-1-en-3-one) dioxygenase (ARD) is found in both prokaryotes and eukaryotes.[4][12][14] ARD enzymes from most species bind ferrous iron and catalyze the oxidation of acireductone to 4-(methylthio)-2-oxobutanoate, the α-keto acid of methionine, and formic acid. However, ARD from Klebsiella oxytoca catalyzes an additional reaction when nickel(II) is bound: it instead produces 3-(methylthio)propionate, formate, and carbon monoxide from the reaction of acireductone with dioxygen. The activity of Fe-ARD is closely interwoven with the methionine salvage pathway, in which the methylthioadenosine product of cellular S-Adenosyl methionine (SAM) reactions is eventually converted to acireductone.
While the exact role of Ni-ARD is not known, it is suspected to help regulate methionine levels by acting as a shunt in the salvage pathway. This K. oxytoca enzyme represents a unique example whereby the metal ion present dictates which reaction is catalyzed. Interestingly, the quercetinases and ARD enzymes all are members of the cupin superfamily, to which the mononuclear iron enzymes also belong.[15] The metal coordination scheme for the QueD enzymes is either a 3-His or 3-His-1-Glu with the exact arrangement being organism-specific.[4] The ARD enzymes all chelate the catalytic metal (either Ni or Fe) through the 3-His-1-Glu motif.[15] In these dioxygenases, the coordinating ligands are provided by both of the typical cupin motifs. In the ARD enzymes, the metal exists in an octahedral arrangement with the three histidine residues comprising a facial triad.[14] The bacterial quercetinase metal centers typically have a trigonal bipyramidal or octahedral coordination environment when there are four protein ligands; the metal centers of the copper-dependent QueD enzymes possesses a distorted tetrahedral geometry in which only the three conserved histidine residues provide coordination ligands.[4][12] Empty coordination sites in all metal centers are occupied by aqua ligands until these are displaced by the incoming substrate.
The ability of these dioxygenases to retain activity in the presence of other metal cofactors with wide ranges of redox potentials suggests the metal center does not play an active role in the activation of dioxygen. Rather, it is thought the metal center functions to hold the substrate in the proper geometry for it to react with dioxygen. In this respect, these enzymes are reminiscent of the intradiol catechol dioxygenases whereby the metal centers activate the substrate for subsequent reaction with dioxygen.
Cofactor-independent dioxygenases

Dioxygenases that catalyze reactions without the need for a cofactor are much more rare in nature than those that do require them. Two dioxygenases, 1H-3-hydroxy-4-oxo-quinoline 2,4-dioxygenase (QDO) and 1H-3-hydroxy-4-oxoquinaldine 2,4-dioxygenase (HDO), have been shown to require neither an organic or metal cofactor.[16] These enzymes catalyze the degradation of quinolone heterocycles in a manner similar to quercetin dioxygenase, but are thought to mediate a radical reaction of a dioxygen molecule with a carbanion on the substrate (figure 5).[17] Both HDO and QDO belong to the α/β hydrolase superfamily of enzymes, although the cataclytic residues in HDO and QDO do not seem to serve the same function as they do in the rest of the enzymes in the α/β hydrolase superfamily.[16]
Clinical significance
Due to the degree of diversity in the dioxygenase family, dioxygenases have a wide range of influences in biology:
- Tryptophan 2,3-dioxygenase (TDO) is important for regulating the levels of tryptophan in the body and is expressed in a high number of human tumors.[18] The other heme iron-dependent dioxygenase, IDO, also has relevance to human health, as it functions in inflammatory responses in the context of certain diseases.[19] Since it affects levels of both tryptophan and kynurenine, IDO has also been implicated in influencing systems related to depression in humans.[20]
- Alkaptonuria is a genetic disease that results in a deficiency of homogentisate 1,2-dioxygenase, which is responsible for catalyzing the formation of 4-maleylacetoacetate from homogentisate.[21] Buildup of homogentisic acid can result in heart valve damage, kidney stones and damage to cartilage in the body.[22]
- Pantothenate kinase-associated neurodegeneration (PKAN) is an autosomal recessive disorder that can lead to the development of iron granules and Lewy bodies in neurons. A study has shown that patients diagnosed with PKAN were found to have increased cysteine levels in the globus pallidus as a consequence of a cysteine dioxygenase deficiency.[23] Patients with PKAN often develop symptoms of dementia and often die at an early age in adulthood.
- In DNA repair, the Fe (II)/2-oxoglutarate-dependent dioxygenase AlkB, functions in the oxidative removal of alkylation damage to DNA. Failure to remove DNA alkylation damage can result in cytotoxicity or mutagenesis during DNA replication.
- Cyclooxygenases (COX), which are responsible for forming prostanoids in the human body, are the target of many NSAID pain relievers.[10] Inhibition of COX leads to reduced inflammation and has an analgesic effect, due to the lowered level of prostaglandin and thromboxane synthesis.
References
- 1 2 Leitgeb, Stefan; Nidetzky, Bernd (1 December 2008). "Structural and functional comparison of 2-His- 1-carboxylate and 3-His metallocentres in non-haem iron(II)-dependent enzymes". Biochemical Society Transactions. 36 (6): 1180. PMID 19021520. doi:10.1042/BST0361180.
- 1 2 3 4 5 6 Abu-Omar, Mahdi M.; Loaiza, Aristobulo; Hontzeas, Nikos (June 2005). "Reaction Mechanisms of Mononuclear Non-Heme Iron Oxygenases". Chemical Reviews. 105 (6): 2227–2252. PMID 15941213. doi:10.1021/cr040653o.
- 1 2 3 4 5 6 Samuel de Visser; Devesh Kumar (2011). Iron-containing enzymes versatile catalysts of hydroxylation reactions in nature. Royal Society of Chemistry. ISBN 978-1-84973-298-7.
- 1 2 3 4 5 6 7 8 Fetzner, S. (27 January 2012). "Ring-Cleaving Dioxygenases with a Cupin Fold". Applied and Environmental Microbiology. 78 (8): 2505–2514. PMC 3318818
 . PMID 22287012. doi:10.1128/AEM.07651-11.
. PMID 22287012. doi:10.1128/AEM.07651-11. - ↑ Stipanuk, Martha H.; Simmons, Chad R.; Andrew Karplus, P.; Dominy, John E. (1 March 2010). "Thiol dioxygenases: unique families of cupin proteins". Amino Acids. 41 (1): 91–102. PMC 3136866 . PMID 20195658. doi:10.1007/s00726-010-0518-2.
- 1 2 Solomon, Edward; Brunold, Thomas; Davis, Mindy; Kemsley, Jyllian; Lee, Sang-Kyu; Lehnert, Nicolai; Neese, Frank; Skulan, Andrew; Yang, Yi-Shan; Zhou, Jing (18 December 1999). "Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes". Chemical Reviews. 100: 235–350. doi:10.1021/cr9900275.
- 1 2 3 Bugg, Timothy DH; Ramaswamy, S (April 2008). "Non-heme iron-dependent dioxygenases: unravelling catalytic mechanisms for complex enzymatic oxidations". Current Opinion in Chemical Biology. 12 (2): 134–140. PMID 18249197. doi:10.1016/j.cbpa.2007.12.007.
- 1 2 Krebs, Carsten; Galonic-Fujimori, Danica; Walsh, Christopher; Bollinger, J. Martin (July 1, 2007). "Non-Heme Fe(IV)–Oxo Intermediates". Accounts of Chemical Research. 40 (7): 484–492. PMC 3870002 . PMID 17542550. doi:10.1021/ar700066p.
- 1 2 3 Efimov, Igor; Basran, Jaswir; Thackray, Sarah J.; Handa, Sandeep; Mowat, Christopher G.; Raven, Emma Lloyd (12 April 2011). "Structure and Reaction Mechanism in the Heme Dioxygenases". Biochemistry. 50 (14): 2717–2724. PMC 3092302 . PMID 21361337. doi:10.1021/bi101732n.
- 1 2 Sono, M; Roach, MP; Coulter, ED; Dawson, JH (Nov 7, 1996). "Heme-Containing Oxygenases.". Chemical Reviews. 96 (7): 2841–2888. PMID 11848843. doi:10.1021/cr9500500.
- 1 2 Thackray, Sarah J.; Mowat, Christopher G.; Chapman, Stephen K. (1 December 2008). "Exploring the mechanism of tryptophan 2,3-dioxygenase". Biochemical Society Transactions. 36 (6): 1120–3. PMC 2652831 . PMID 19021508. doi:10.1042/BST0361120.
- 1 2 3 4 Schaab, MR; Barney, BM; Francisco, WA (Jan 24, 2006). "Kinetic and spectroscopic studies on the quercetin 2,3-dioxygenase from Bacillus subtilis.". Biochemistry. 45 (3): 1009–16. PMID 16411777. doi:10.1021/bi051571c.
- ↑ "The Single Superoxide Dismutase of Rhodobacter capsulatus Is a Cambialistic, Manganese-Containing Enzyme". Jb.asm.org. Retrieved 2014-03-11.
- 1 2 Maroney, Michael J.; Ciurli, Stefano (26 December 2013). "Nonredox Nickel Enzymes". Chemical Reviews. 114 (8): 4206–28. PMID 24369791. doi:10.1021/cr4004488.
- 1 2 Boer, Jodi L.; Mulrooney, Scott B.; Hausinger, Robert P. (February 2014). "Nickel-dependent metalloenzymes". Archives of Biochemistry and Biophysics. 544: 142–152. PMC 3946514 . PMID 24036122. doi:10.1016/j.abb.2013.09.002.
- 1 2 S., Fetzner (1 November 2002). "Oxygenases without requirement for cofactors or metal ions". Applied Microbiology and Biotechnology. 60 (3): 243–257. PMID 12436305. doi:10.1007/s00253-002-1123-4.
- ↑ Bugg, Timothy D.H. (September 2003). "Dioxygenase enzymes: catalytic mechanisms and chemical models". Tetrahedron. 59 (36): 7075–7101. doi:10.1016/S0040-4020(03)00944-X.
- ↑ Pilotte, L.; Larrieu, P.; Stroobant, V.; Colau, D.; Dolusic, E.; Frederick, R.; De Plaen, E.; Uyttenhove, C.; Wouters, J.; Masereel, B.; Van den Eynde, B. J. (30 January 2012). "Reversal of tumoral immune resistance by inhibition of tryptophan 2,3-dioxygenase". Proceedings of the National Academy of Sciences. 109 (7): 2497–2502. PMC 3289319 . PMID 22308364. doi:10.1073/pnas.1113873109.
- ↑ Murakami, Yuki; Hoshi, Masato; Imamura, Yukio; Arioka, Yuko; Yamamoto, Yasuko; Saito, Kuniaki (2013). "Remarkable Role of Indoleamine 2,3-Dioxygenase and Tryptophan Metabolites in Infectious Diseases: Potential Role in Macrophage-Mediated Inflammatory Diseases". Mediators of Inflammation. 2013: 1–9. doi:10.1155/2013/391984.
- ↑ Sublette, M. E.; Postolache, T. T. (24 August 2012). "Neuroinflammation and Depression: The Role of Indoleamine 2,3-dioxygenase (IDO) as a Molecular Pathway". Psychosomatic Medicine. 74 (7): 668–672. PMID 22923699. doi:10.1097/PSY.0b013e318268de9f.
- ↑ Voet, Donald Voet, Judith G. (2011). Biochemistry (4th ed.). Hoboken, NJ: John Wiley & Sons. p. 1045. ISBN 0470917458.
- ↑ Phornphutkul, Chanika; Introne, Wendy J.; Perry, Monique B.; Bernardini, Isa; Murphey, Mark D.; Fitzpatrick, Diana L.; Anderson, Paul D.; Huizing, Marjan; Anikster, Yair; Gerber, Lynn H.; Gahl, William A. (26 December 2002). "Natural History of Alkaptonuria". New England Journal of Medicine. 347 (26): 2111–2121. PMID 12501223. doi:10.1056/NEJMoa021736.
- ↑ Perry, TL; Kish SJ; Norman MG; Yong VW; Whiting S; Crichton JU; Hansen S (1985). "Hallervorden-Spatz disease: cysteine accumulation and cysteine dioxygenase deficiency in the globus pallidus". Ann. Neurol. 18 (4): 482–489. PMID 4073841. doi:10.1002/ana.410180411.