Coot (software)
|
the Coot main window (version 0.5pre) | |
| Developer(s) |
Paul Emsley Kevin D. Cowtan |
|---|---|
| Initial release | 2002 |
| Stable release |
0.8.7
/ November 8, 2016 |
| Operating system | Windows, Linux, OS X, Unix |
| Type | Molecular modelling |
| License | GNU General Public License |
| Website |
http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot http://www.biop.ox.ac.uk/coot/ |

The program Coot (Crystallographic Object-Oriented Toolkit)[1][2] is used to display and manipulate atomic models of macromolecules, typically of proteins or nucleic acids, using 3D computer graphics. It is primarily focused on building and validation of atomic models into three-dimensional electron density maps obtained by X-ray crystallography methods, although it has also been applied to data from electron microscopy.
Overview
Coot displays electron density maps and atomic models and allows model manipulations such as idealization, real space refinement, manual rotation/translation, rigid-body fitting, ligand search, solvation, mutations, rotamers, and Ramachandran idealization. The software is designed to be easy-to-learn for novice users, achieved by ensuring that tools for common tasks are 'discoverable' through familiar user interface elements (menus and toolbars), or by intuitive behaviour (mouse controls). Recent developments have enhanced the usability of the software for expert users, with customisable key bindings, extensions, and an extensive scripting interface.
Coot is free software, distributed under the GNU GPL. It is available from the Coot web site[3] originally at the University of York, and now at the MRC Laboratory of Molecular Biology. Pre-compiled binaries are also available for Linux and Windows from the web page and CCP4, and for Mac OS X through Fink and CCP4. Additional support is available through the Coot wiki and an active COOT mailing list.[4][5]
The primary author is Paul Emsley (MRC-LMB at Cambridge). Other contributors include Kevin Cowtan, Bernhard Lohkamp and Stuart McNicholas (University of York), William Scott (University of California at Santa Cruz), and Eugene Krissinel (Daresbury Laboratory).
Features
Coot can be used to read files containing 3D atomic coordinate models of macromolecular structures in a number of formats, including pdb, mmcif, and Shelx files. The model may then be rotated in 3D and viewed from any viewpoint. The atomic model is represented by default using a stick-model, with vectors representing chemical bonds. The two halves of each bond are coloured according to the element of the atom at that end of the bond, allowing chemical structure and identity to be visualised in a manner familiar to most chemists.
Coot can also display electron density, which is the result of structure determination experiments such as X-ray crystallography and EM reconstruction. The density is contoured using a 3D-mesh. The contour level controlled using the mouse wheel for easy manipulation - this provides a simple way for the user to get an idea of the 3D electron density profile without the visual clutter of multiple contour levels. Electron density may be read into the program from ccp4 or cns map formats, though it is more common to calculate an electron density map directly from the X-ray diffraction data, read from an mtz, hkl, fcf or mmcif file.
Coot provides extensive features for model building and refinement (i.e. adjusting the model to better fit the electron density), and for validation (i.e. checking that the atomic model agrees with the experimentally derived electron density and makes chemical sense). The most important of these tools is the real space refinement engine, which will optimize the fit of a section of atomic model to the electron density in real time, with graphical feedback. The user may also intervene in this process, dragging the atoms into the right places if the initial model is too far away from the corresponding electron density.
Model building tools
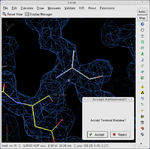
Tools for general model building:
- C-alpha baton mode - trace the main chain of a protein by placing correctly spaced alpha-carbon atoms.
- Ca Zone -> Mainchain - convert an initial trace of the alpha-carbon atoms to a full main-chain trace.
- Place helix here - fit a sequence of amino acids in alpha helix conformation into density.
- Place strand here - fit a sequence of amino acids in beta strand conformation into density.
- Ideal DNA/RNA - build an ideal DNA or RNA fragment.
- Find ligands - find and fit a model to any small molecule which may be bound to the macromolecule.
Tools for moving existing atoms:
- Real space refine zone - optimize the fit of the model to the electron density, while preserving stereochemistry.
- Regularize zone - optimize stereochemistry.
- Rigid body fit zone - optimize the fit of a rigid body to the electron density.
- Rotate/translate zone - manually position a rigid body.
- Rotamer tools (auto fit rotamer, manual rotamer, mutate and autofit, simple mutate)
- Torsion editing (edit chi angles, edit main chain torsions, general torsions)
- Other protein tools (flip peptide, flip sidechain, cis <-> trans)
Tools for adding atoms to the model:
- Find waters - add ordered solvent molecules to the model
- Add terminal residue - extend a protein or nucleotide chain
- Add alternate conformation
- Place atom at pointer
Validation tools

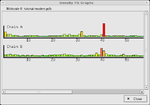
In macromolecular crystallography, the observed data is often weak and the observation-to-parameter ratio near 1. As a result, it is possible to build an incorrect atomic model into the electron density in some cases. To avoid this, careful validation is required. Coot provides a range of validation tools, listed below. Having built an initial model, it is usual to check all of these and reconsider any parts of the model which are highlighted as problematic before deposition of the atomic coordinates with a public database.
- Ramachandran plot - validate the torsion angles of a protein chain.
- Kleywegt plot - examine differences between the torsions of NCS-related chains.
- Incorrect chiral volumes - check for chiral centres with the wrong handedness.
- Unmodelled blobs - check for electron density not accounted for by existing atoms.
- Difference map peaks - check for large differences between observed and calculated density.
- Check/Delete waters - check for water molecules which do not fit the density.
- Check waters by difference map variance
- Geometry analysis - check for improbable bond lengths, angles, etc.
- Peptide omega analysis - check for non-planar peptide bonds.
- Temperature factor variance analysis -
- GLN and ASN B-factor outliers -
- Rotamer analysis - check for unusual protein side-chain conformations.
- Density fit analysis - identify parts of the model which don't fit the density.
- Probe clashes - check for Hydrogen atoms with inappropriate environments (using Molprobity).
- NCS differences - check for general differences between NCS related chains.
- Pukka puckers - check for unusual DNA/RNA conformations.
Program architecture

Coot is built upon a number of libraries. Crystallographic tools include the Clipper library[6] for manipulating electron density and providing crystallographic algorithms, and the MMDB[7] for the manipulation of atomic models. Other dependencies include FFTW, and the GNU Scientific Library.
Much of the program's functionality is available through a scripting interface, which provides access from both the Python and Guile scripting languages.
Relation to CCP4mg
The CCP4mg molecular graphics software[8][9] from Collaborative Computational Project Number 4 is a related project with which Coot shares some code. The projects are focused on slightly different problems, with CCP4mg dealing with presentation graphics and movies, whereas Coot deals with model building and validation.
Impact in the crystallographic computing community
The software has gained considerable popularity over the past 5 years, overtaking widely used packages such as 'O',[10] XtalView,[11] and Turbo Frodo.[12] The primary publication has been cited in over 10,000 independent scientific papers since 2004.[13]
References
- ↑ P. Emsley; B. Lohkamp; W.G. Scott; Cowtan (2010). "Features and Development of Coot". Acta Crystallographica. D66: 486–501.
- ↑ P. Emsley; K. Cowtan (2004). "Coot: model-building tools for molecular graphics". Acta Crystallographica. D60: 2126–2132.
- ↑ "Coot". Mrc-lmb.cam.ac.uk. Retrieved 2017-02-27.
- ↑ "Coot - CCP4 wiki". Strucbio.biologie.uni-konstanz.de. Retrieved 2017-02-27.
- ↑ "Coot List At Www.Jiscmail.Ac.Uk". JISCMail. Retrieved 2017-02-27.
- ↑ "Dr Kevin Cowtan - About staff, The University of York". Ysbl.york.ac.uk. 2014-10-23. Retrieved 2017-02-27.
- ↑
- ↑ L. Potterton, S. McNicholas, E. Krissinel, J. Gruber, K. Cowtan, P. Emsley, G. N. Murshudov, S. Cohen, A. Perrakis and M. Noble (2004). "Developments in the CCP4 molecular-graphics project". Acta Crystallogr. D60: 2288–2294.
- ↑
- ↑ "Home Page of Alwyn Jones". Xray.bmc.uu.se. Retrieved 2017-02-27.
- ↑ "CCMS Software - XtalView". Sdsc.edu. 2006-08-09. Retrieved 2017-02-27.
- ↑ "Turbo Frodo Description". Csb.yale.edu. 1999-03-26. Retrieved 2017-02-27.
- ↑ "Coot model building tools for molecular graphics - Google Scholar". Scholar.google.co.uk. Retrieved 2017-02-27.