Combined bisulfite restriction analysis
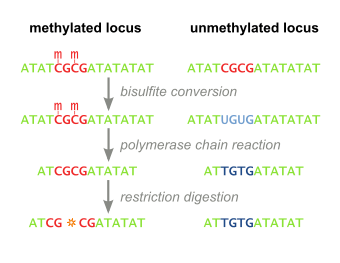
Combined Bisulfite Restriction Analysis (or COBRA) is a molecular biology technique that allows for the sensitive quantification of DNA methylation levels at a specific genomic locus on a DNA sequence in a small sample of genomic DNA.[1] The technique is a variation of bisulfite sequencing, and combines bisulfite conversion based polymerase chain reaction with restriction digestion. Originally developed to reliably handle minute amounts of genomic DNA from microdissected paraffin-embedded tissue samples, the technique has since seen widespread usage in cancer research and epigenetics studies.[2]
Procedure
Bisulfite Treatment
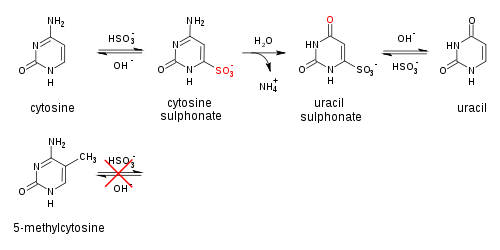
Genomic DNA of interest is treated with sodium bisulfite, which introduces methylation-dependent sequence differences. During sodium bisulfite treatment, unmethylated cytosine residues are converted to uracil, while methylated cytosine residues are unaffected.
PCR Amplification
Bisulfite treated DNA is then PCR amplified, resulting in cytosine residues at originally methylated positions, and thymine residues at originally unmethylated position (that were converted to uracil). Primers used during this step do not contain CpG sites (the common target of cytosine methylation), so the amplificiation process does not discriminate between templates based on methylation status. PCR products are purified to ensure complete digestion in the following step.
Restriction Digest
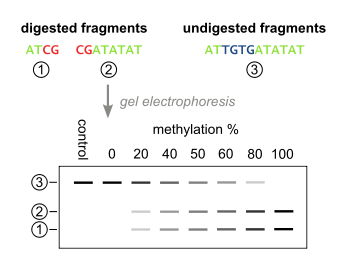
The above steps lead to the methylation dependent retention or loss of CpG-containing restriction enzyme sites, such as those for TaqI (TCGA) and BstUI (CGCG), depending on whether the cytosine residue was originally methylated or not, respectively. Due to the methylation-independent amplification in the above step, the resulting PCR products will be a mixed population of fragments that have lost or retained CpG-containing restriction enzyme sites, whose respective percentages will be directly correlated to the original level of DNA methylation in the sample DNA.
PCR products are then treated with a restriction enzyme (e.g. BstUI), which will only cleave sites that were originally methylated (CGCG), while leaving sites that were originally unmethylated (TGTG). To ensure that all CpG sites are retained due to originally being methylated, and not a remnant of incomplete bisulfite conversion, a control digestion is performed, with enzymes such as Hsp92II which recognizes the sequence CATG, none of which should be remaining after bisulfite conversion (with the rare exception of non-CpG methylation) and thus no cleavage should occur if bisulfite conversion was complete.
Quantification
The digested fragments are then separated by polyacrylamide gel electrophoresis with the expected appearance of bands corresponding to a single large undigested fragment, and multiple smaller bands corresponding to digested fragments. Quantitative amount of DNA in these bands can be determined with a device such as a phosphoimager, after which the methylation percentage of the original sample can be calculated by:

Usage and Applications
COBRA has been used extensively in many research-based applications such as screening for DNA methylation changes at gene promoters in cancer studies,[3][4] detecting altered methylation patterns at imprinted genes,[5] and characterizing methylation patterns in the genome during development in mammals.[6][7]
In medicine, COBRA has been used as a tool to help diagnose human disease involving aberrant DNA methylation. Researchers utilized COBRA in conjunction with denaturing high performance liquid chromatography in the diagnosis of the genetic imprinting disorder Russell-Silver syndrome where hypomethylation of the imprinted gene H19 is responsible for the disorder in up to 50% of patients.[8]
Strengths
- Simple, fast and inexpensive: In COBRA, DNA methylation levels are easily and quickly measured without the need for laborious sub-cloning and sequencing, as with bisulfite sequencing. The assay is straightforward and can be done with standard inexpensive molecular biology reagents.
- High compatibility: Due to the PCR and purification steps, the method not only works with very small amounts of genomic DNA, but also samples that have been treated with paraffin, both of which can be problems in other DNA methylation quantification protocols such as Southern blotting and methylation-sensitive restriction enzyme digestion followed by PCR.
- Quantitative: This is in contrast to methylation-specific PCR, which is qualitative. With COBRA, DNA methylation levels can be directly quantified at a given locus, yielding more information per assay.
- Scalability for high-throughput sample processing: With COBRA, many regions of interest can be processed in parallel in separate samples digested with the same restriction enzyme. This is in contrast to bisulfite sequencing analysis, where each region needs to be examined rigorously by sequencing many clones per locus, costing more time.
- Multiple queries per assay: Methylation status can be interrogated at multiple CpG-containing restriction sites in a single digestion assay.
Weaknesses
- The assay is limited to using existing restriction sites in the region of interest, and methylation that does not occur in the context of a specific restriction site will not be assayed.
- Incomplete digestion by restriction enzymes after PCR can confound the analysis: incomplete digestion would suggest lack of DNA methylation (if cutting with a methylation-sensitive enzyme such as HpaII). It is also known that BstUI can cut at unconverted sites, leading to overestimation of methylation levels and so the use of HpaII is often needed.[9]
- In complex samples, cell-type heterogeneity can confound the analysis since the DNA is not being sequenced, heterogeneity in sequences from different cells in the sample (i.e. different cell populations within a tumor) that have acquired mutations in the interrogated region, such as changing the CG dinucleotide to CA or CT, would result in loss of the restriction site giving rise to an apparently methylated region due to lack of digestion. This would skew the quantification of DNA methylation levels in a given sample.
Alternatives
In general, COBRA is often combined with other DNA methylation analyses and is frequently used in the initial screening of a loci of interest. If COBRA suggests altered methylation patterns, then more rigorous, labor-intensive techniques can be applied, such as bisulfite sequencing or MeDIP.
References
- ↑ Xiong, Zhenggang; Laird, Peter W. (1997). "COBRA: a sensitive and quantitative DNA methylation assay". Nucleic Acids Research. 25 (12): 2532–2534. doi:10.1093/nar/25.12.2532.
- ↑ Laird, Peter W. (2003). "The power and the promise of DNA methylation markers". Nature Reviews Cancer. 3 (April): 253–266. PMID 12671664. doi:10.1038/nrc1045. This paper had 576 references as of February 2–11, according to Scopus
- ↑ Shen, Lanlan; et al. (2005). "MGMT Promoter Methylation and Field Defect in Sporadic Colorectal Cancer". Journal of the National Cancer Institute. 97 (18): 1330–1338. PMID 16174854. doi:10.1093/jnci/dji275.
- ↑ Suter, Catherine, M.; et al. (2004). "Germline epimutation in MLH1 in individuals with multiple cancers". Nature Genetics. 36 (5): 497–501. doi:10.1038/ng1342.
- ↑ Bliek, Jet; et al. (2009). "Hypomethylation at multiple maternally methylated imprinted regions including PLAGL1 and GNAS loci in Beckwith–Wiedemann_syndrome, and characterizing DNA methylation patterns in the genome during development in mammals". European Journal of Human Genetics. 17 (5): 611–618. doi:10.1038/ejhg.2008.233.
- ↑ Wernig, Marius; et al. (2007). "In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state". Nature. 448 (7151): 318–324. PMID 17554336. doi:10.1038/nature05944.
- ↑ Mikkelsen, Tarjei S.; et al. (2008). "Dissecting direct reprogramming through integrative genomic analysis". Nature. 454 (July): 49–56. PMC 2754827
 . PMID 18509334. doi:10.1038/nature07056.
. PMID 18509334. doi:10.1038/nature07056. - ↑ Hattori, M.; et al. (2009). "Diagnosis of Russell-Silver syndrome by the combined bisulfite restriction analysis-denaturing high-performance liquid chromatography assay". Genetic testing and molecular biomarkers. 13 (5): 623–630. doi:10.1089/gtmb.2009.0018.
- ↑ Fraga, Mario F.; Esteller, Manel (2002). "DNA Methylation: A Profile of Methods and Applications". Biotechniques. 33: 633–649.