Cofactor (biochemistry)

A cofactor is a non-protein chemical compound or metallic ion that is required for a protein's biological activity to happen. These proteins are commonly enzymes, and cofactors can be considered "helper molecules" that assist in biochemical transformations. The rates at which this happen are characterized by enzyme kinetics.
Cofactors can be subclassified as either inorganic ions or complex organic molecules called coenzymes,[1] the latter of which is mostly derived from vitamins and other organic essential nutrients in small amounts. A coenzyme that is tightly or even covalently bound is termed a prosthetic group.[2] Cosubstrates are transiently bound to the protein and will be released at some point, then get back in. The prosthetic groups, on the other hand, are bound permanently to the protein. Both of them have the same function, which is to facilitate the reaction of enzymes and protein. Additionally, some sources also limit the use of the term "cofactor" to inorganic substances.[3][4] An inactive enzyme without the cofactor is called an apoenzyme, while the complete enzyme with cofactor is called a holoenzyme.[5]
Some enzymes or enzyme complexes require several cofactors. For example, the multienzyme complex pyruvate dehydrogenase[6] at the junction of glycolysis and the citric acid cycle requires five organic cofactors and one metal ion: loosely bound thiamine pyrophosphate (TPP), covalently bound lipoamide and flavin adenine dinucleotide (FAD), and the cosubstrates nicotinamide adenine dinucleotide (NAD+) and coenzyme A (CoA), and a metal ion (Mg2+).[7]
Organic cofactors are often vitamins or made from vitamins. Many contain the nucleotide adenosine monophosphate (AMP) as part of their structures, such as ATP, coenzyme A, FAD, and NAD+. This common structure may reflect a common evolutionary origin as part of ribozymes in an ancient RNA world. It has been suggested that the AMP part of the molecule can be considered to be a kind of "handle" by which the enzyme can "grasp" the coenzyme to switch it between different catalytic centers.[8]
Classification
Cofactors can be divided into two broad groups: organic cofactors, such as flavin or heme, and inorganic cofactors, such as the metal ions Mg2+, Cu+, Mn2+, or iron-sulfur clusters.
Organic cofactors are sometimes further divided into coenzymes and prosthetic groups. The term coenzyme refers specifically to enzymes and, as such, to the functional properties of a protein. On the other hand, "prosthetic group" emphasizes the nature of the binding of a cofactor to a protein (tight or covalent) and, thus, refers to a structural property. Different sources give slightly different definitions of coenzymes, cofactors, and prosthetic groups. Some consider tightly bound organic molecules as prosthetic groups and not as coenzymes, while others define all non-protein organic molecules needed for enzyme activity as coenzymes, and classify those that are tightly bound as coenzyme prosthetic groups. It should be noted that these terms are often used loosely.
A 1979 letter in Trends in Biochemical Sciences noted the confusion in the literature and the essentially arbitrary distinction made between prosthetic groups and coenzymes and proposed the following scheme. Here, cofactors were defined as an additional substance apart from protein and substrate that is required for enzyme activity and a prosthetic group as a substance that undergoes its whole catalytic cycle attached to a single enzyme molecule. However, the author could not arrive at a single all-encompassing definition of a "coenzyme" and proposed that this term be dropped from use in the literature.[9]
Inorganic
Metal ions
Metal ions are common cofactors.[10] The study of these cofactors falls under the area of bioinorganic chemistry. In nutrition, the list of essential trace elements reflects their role as cofactors. In humans this list commonly includes iron, magnesium, manganese, cobalt, copper, zinc, and molybdenum.[11] Although chromium deficiency causes impaired glucose tolerance, no human enzyme that uses this metal as a cofactor has been identified.[12][13] Iodine is also an essential trace element, but this element is used as part of the structure of thyroid hormones rather than as an enzyme cofactor.[14] Calcium is another special case, in that it is required as a component of the human diet, and it is needed for the full activity of many enzymes, such as nitric oxide synthase, protein phosphatases, and adenylate kinase, but calcium activates these enzymes in allosteric regulation, often binding to these enzymes in a complex with calmodulin.[15] Calcium is, therefore, a cell signaling molecule, and not usually considered a cofactor of the enzymes it regulates.[16]
Other organisms require additional metals as enzyme cofactors, such as vanadium in the nitrogenase of the nitrogen-fixing bacteria of the genus Azotobacter,[17] tungsten in the aldehyde ferredoxin oxidoreductase of the thermophilic archaean Pyrococcus furiosus,[18] and even cadmium in the carbonic anhydrase from the marine diatom Thalassiosira weissflogii.[19][20]
In many cases, the cofactor includes both an inorganic and organic component. One diverse set of examples is the heme proteins, which consist of a porphyrin ring coordinated to iron.[21]

Iron-sulfur clusters
Iron-sulfur clusters are complexes of iron and sulfur atoms held within proteins by cysteinyl residues. They play both structural and functional roles, including electron transfer, redox sensing, and as structural modules.[22]
Organic
Organic cofactors are small organic molecules (typically a molecular mass less than 1000 Da) that can be either loosely or tightly bound to the enzyme and directly participate in the reaction.[5][23][24][25] In the latter case, when it is difficult to remove without denaturing the enzyme, it can be called a prosthetic group. It is important to emphasize that there is no sharp division between loosely and tightly bound cofactors.[5] Indeed, many such as NAD+ can be tightly bound in some enzymes, while it is loosely bound in others.[5] Another example is thiamine pyrophosphate (TPP), which is tightly bound in transketolase or pyruvate decarboxylase, while it is less tightly bound in pyruvate dehydrogenase.[26] Other coenzymes, flavin adenine dinucleotide (FAD), biotin, and lipoamide, for instance, are covalently bound. Tightly bound cofactors are, in general, regenerated during the same reaction cycle, while loosely bound cofactors can be regenerated in a subsequent reaction catalyzed by a different enzyme. In the latter case, the cofactor can also be considered a substrate or cosubstrate.
Vitamins can serve as precursors to many organic cofactors (e.g., vitamins B1, B2, B6, B12, niacin, folic acid) or as coenzymes themselves (e.g., vitamin C). However, vitamins do have other functions in the body.[27] Many organic cofactors also contain a nucleotide, such as the electron carriers NAD and FAD, and coenzyme A, which carries acyl groups. Most of these cofactors are found in a huge variety of species, and some are universal to all forms of life. An exception to this wide distribution is a group of unique cofactors that evolved in methanogens, which are restricted to this group of archaea.[28]
Vitamins and derivatives
Non-vitamins
Cofactors as metabolic intermediates
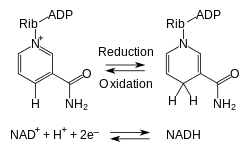
Metabolism involves a vast array of chemical reactions, but most fall under a few basic types of reactions that involve the transfer of functional groups.[58] This common chemistry allows cells to use a small set of metabolic intermediates to carry chemical groups between different reactions.[59] These group-transfer intermediates are the loosely bound organic cofactors, often called coenzymes.
Each class of group-transfer reaction is carried out by a particular cofactor, which is the substrate for a set of enzymes that produce it, and a set of enzymes that consume it. An example of this are the dehydrogenases that use nicotinamide adenine dinucleotide (NAD+) as a cofactor. Here, hundreds of separate types of enzymes remove electrons from their substrates and reduce NAD+ to NADH. This reduced cofactor is then a substrate for any of the reductases in the cell that require electrons to reduce their substrates.[30]
Therefore, these cofactors are continuously recycled as part of metabolism. As an example, the total quantity of ATP in the human body is about 0.1 mole. This ATP is constantly being broken down into ADP, and then converted back into ATP. Thus, at any given time, the total amount of ATP + ADP remains fairly constant. The energy used by human cells requires the hydrolysis of 100 to 150 moles of ATP daily, which is around 50 to 75 kg. In typical situations, humans use up their body weight of ATP over the course of the day.[60] This means that each ATP molecule is recycled 1000 to 1500 times daily.
Evolution
Organic cofactors, such as ATP and NADH, are present in all known forms of life and form a core part of metabolism. Such universal conservation indicates that these molecules evolved very early in the development of living things.[61] At least some of the current set of cofactors may, therefore, have been present in the last universal ancestor, which lived about 4 billion years ago.[62][63]
Organic cofactors may have been present even earlier in the history of life on Earth.[64] The nucleotide adenosine is present in cofactors that catalyse many basic metabolic reactions such as methyl, acyl, and phosphoryl group transfer, as well as redox reactions. This ubiquitous chemical scaffold has, therefore, been proposed to be a remnant of the RNA world, with early ribozymes evolving to bind a restricted set of nucleotides and related compounds.[65][66] Adenosine-based cofactors are thought to have acted as interchangeable adaptors that allowed enzymes and ribozymes to bind new cofactors through small modifications in existing adenosine-binding domains, which had originally evolved to bind a different cofactor.[8] This process of adapting a pre-evolved structure for a novel use is known as exaptation.
A computational method, IPRO, recently predicted mutations that experimentally switched the cofactor specificity of Candida boidinii xylose reductase from NADPH to NADH.[67]
History
The first organic cofactor to be discovered was NAD+, which was identified by Arthur Harden and William Youndin 1906.[68] They noticed that adding boiled and filtered yeast extract greatly accelerated alcoholic fermentation in unboiled yeast extracts. They called the unidentified factor responsible for this effect a coferment. Through a long and difficult purification from yeast extracts, this heat-stable factor was identified as a nucleotide sugar phosphate by Hans von Euler-Chelpin.[69] Other cofactors were identified throughout the early 20th century, with ATP being isolated in 1929 by Karl Lohmann,[70] and coenzyme A being discovered in 1945 by Fritz Albert Lipmann.[71]
The functions of these molecules were at first mysterious, but, in 1936, Otto Heinrich Warburg identified the function of NAD+ in hydride transfer.[72] This discovery was followed in the early 1940s by the work of Herman Kalckar, who established the link between the oxidation of sugars and the generation of ATP.[73] This confirmed the central role of ATP in energy transfer that had been proposed by Fritz Albert Lipmann in 1941.[74] Later, in 1949, Morris Friedkin and Albert L. Lehninger proved that NAD+ linked metabolic pathways such as the citric acid cycle and the synthesis of ATP.[75]
Protein-derived cofactors
In a number of enzymes, the moiety that acts as a cofactor is formed by post-translational modification of a part of the protein sequence. This often replaces the need for an external binding factor, such as a metal ion, for protein function. Potential modifications could be oxidation of aromatic residues, binding between residues, cleavage or ring-forming.[76] These alterations are distinct from other post-translation protein modifications, such as phosphorylation, methylation, or glycosylation in that the amino acids typically acquire new functions. This increases the functionality of the protein; unmodified amino acids are typically limited to acid-base reactions, and the alteration of resides can give the protein electrophilic sites or the ability to stabilize free radicals.[76] Examples of cofactor production include tryptophan tryptophylquinone (TTQ), derived from two tryptophan side chains,[77] and 4-methylidene-imidazole-5-one (MIO), derived from an Ala-Ser-Gly motif.[78] Characterization of protein-derived cofactors is conducted using X-ray crystallography and mass spectroscopy; structural data is necessary because sequencing does not readily identify the altered sites.
Non-enzymatic cofactors
The term is used in other areas of biology to refer more broadly to non-protein (or even protein) molecules that either activate, inhibit, or are required for the protein to function. For example, ligands such as hormones that bind to and activate receptor proteins are termed cofactors or coactivators, whereas molecules that inhibit receptor proteins are termed corepressors. One such example is the G protein-coupled receptor family of receptors, which are frequently found in sensory neurons. Ligand binding to the receptors activates the G protein, which then activates an enzyme to activate the effector.[79] In order to avoid confusion, it has been suggested that such proteins that have ligand-binding mediated activation or repression be referred to as coregulators.[80]
See also
- Enzyme catalysis
- Inorganic chemistry
- Organometallic chemistry
- Bioorganometallic chemistry
- Cofactor engineering
References
- ↑ Hasim, Onn (2010). Coenzyme, Cofactor and Prosthetic Group - Ambiguous Biochemical Jargon. Kuala Lumpur: Biochemical Education. pp. 93–94.
- ↑ Nelson, David (2008). <Lehninger Principles of Biochemistry>. New York: W.H. Freeman and Company. p. 184.
- ↑ "coenzymes and cofactors". Retrieved 2007-11-17.
- ↑ "Enzyme Cofactors". Archived from the original on 2003-05-05. Retrieved 2007-11-17.
- 1 2 3 4 5 6 Sauke, David J.; Metzler, David E.; Metzler, Carol M. (2001). Biochemistry: the chemical reactions of living cells (2nd ed.). San Diego: Harcourt/Academic Press. ISBN 0-12-492540-5.
- ↑ Jordan, Frank; Patel, Mulchand S. (2004). Thiamine: catalytic mechanisms in normal and disease states. New York, N.Y: Marcel Dekker. p. 588. ISBN 0-8247-4062-9.
- ↑ "Pyruvate Dehydrogenase Complex". Chemistry LibreTexts. 2013-10-02. Retrieved 2017-05-10.
- 1 2 Denessiouk KA, Rantanen VV, Johnson MS (2001). "Adenine recognition: a motif present in ATP-, CoA-, NAD-, NADP-, and FAD-dependent proteins". Proteins. 44 (3): 282–91. PMID 11455601. doi:10.1002/prot.1093.
- ↑ Bryce CFA (March 1979). "SAM – semantics and misunderstandings". Trends Biochem. Sci. 4 (3): N62. doi:10.1016/0968-0004(79)90255-X.
- ↑ Metal Ions in Life Sciences
- ↑ Aggett PJ (1985). "Physiology and metabolism of essential trace elements: an outline". Clin Endocrinol Metab. 14 (3): 513–43. PMID 3905079. doi:10.1016/S0300-595X(85)80005-0.
- ↑ Stearns DM (2000). "Is chromium a trace essential metal?". BioFactors. 11 (3): 149–62. PMID 10875302. doi:10.1002/biof.5520110301.
- ↑ Vincent JB (1 April 2000). "The biochemistry of chromium". J. Nutr. 130 (4): 715–8. PMID 10736319.
- ↑ Cavalieri RR (1997). "Iodine metabolism and thyroid physiology: current concepts". Thyroid. 7 (2): 177–81. PMID 9133680. doi:10.1089/thy.1997.7.177.
- ↑ Clapham DE (2007). "Calcium signaling". Cell. 131 (6): 1047–58. PMID 18083096. doi:10.1016/j.cell.2007.11.028.
- ↑ Niki I, Yokokura H, Sudo T, Kato M, Hidaka H (1996). "Ca2+ signaling and intracellular Ca2+ binding proteins". J. Biochem. 120 (4): 685–98. PMID 8947828. doi:10.1093/oxfordjournals.jbchem.a021466.
- ↑ Eady RR (1988). "The vanadium-containing nitrogenase of Azotobacter". BioFactors. 1 (2): 111–6. PMID 3076437.
- ↑ Chan MK, Mukund S, Kletzin A, Adams MW, Rees DC (1995). "Structure of a hyperthermophilic tungstopterin enzyme, aldehyde ferredoxin oxidoreductase". Science. 267 (5203): 1463–9. PMID 7878465. doi:10.1126/science.7878465.
- ↑ Lane TW, Morel FM (2000). "A biological function for cadmium in marine diatoms". Proc. Natl. Acad. Sci. U.S.A. 97 (9): 4627–31. PMC 18283
 . PMID 10781068. doi:10.1073/pnas.090091397.
. PMID 10781068. doi:10.1073/pnas.090091397. - ↑ Lane TW, Saito MA, George GN, Pickering IJ, Prince RC, Morel FM (2005). "Biochemistry: a cadmium enzyme from a marine diatom". Nature. 435 (7038): 42. PMID 15875011. doi:10.1038/435042a.
- ↑ Li, Ting; Bonkovsky, Herbert L.; Guo, Jun-tao (2011-01-01). "Structural analysis of heme proteins: implications for design and prediction". BMC Structural Biology. 11: 13. ISSN 1472-6807. PMC 3059290 . PMID 21371326. doi:10.1186/1472-6807-11-13.
- ↑ Meyer J (February 2008). "Iron-sulfur protein folds, iron-sulfur chemistry, and evolution". J. Biol. Inorg. Chem. 13 (2): 157–70. PMID 17992543. doi:10.1007/s00775-007-0318-7.
- ↑ Palmer, Trevor (1981). Understanding enzymes. New York: Horwood. ISBN 0-85312-307-1.
- ↑ Cox, Michael; Lehninger, Albert L; Nelson, David R. (2000). Lehninger principles of biochemistry (3rd ed.). New York: Worth Publishers. ISBN 1-57259-153-6.
- ↑ Farrell, Shawn O.; Campbell, Mary K. (2009). Biochemistry (6th ed.). Pacific Grove: Brooks Cole. ISBN 0-495-39041-0.
- ↑ Morey, A. V.; Juni, Elliot (1968-06-10). "Studies on the Nature of the Binding of Thiamine Pyrophosphate to Enzymes". Journal of Biological Chemistry. 243 (11): 3009–3019. ISSN 0021-9258. PMID 4968184.
- ↑ Bolander FF (2006). "Vitamins: not just for enzymes". Curr Opin Investig Drugs. 7 (10): 912–5. PMID 17086936.
- ↑ Rouvière PE, Wolfe RS (15 June 1988). "Novel biochemistry of methanogenesis". J. Biol. Chem. 263 (17): 7913–6. PMID 3131330.
- ↑ Frank RA, Leeper FJ, Luisi BF (2007). "Structure, mechanism and catalytic duality of thiamine-dependent enzymes". Cell. Mol. Life Sci. 64 (7–8): 892–905. PMID 17429582. doi:10.1007/s00018-007-6423-5.
- 1 2 Pollak N, Dölle C, Ziegler M (2007). "The power to reduce: pyridine nucleotides—small molecules with a multitude of functions". Biochem. J. 402 (2): 205–18. PMC 1798440 . PMID 17295611. doi:10.1042/BJ20061638.
- ↑ Eliot AC, Kirsch JF (2004). "Pyridoxal phosphate enzymes: mechanistic, structural, and evolutionary considerations". Annu. Rev. Biochem. 73: 383–415. PMID 15189147. doi:10.1146/annurev.biochem.73.011303.074021.
- ↑ Banerjee R, Ragsdale SW (2003). "The many faces of vitamin B12: catalysis by cobalamin-dependent enzymes". Annu. Rev. Biochem. 72: 209–47. PMID 14527323. doi:10.1146/annurev.biochem.72.121801.161828.
- ↑ Jitrapakdee S, Wallace JC (2003). "The biotin enzyme family: conserved structural motifs and domain rearrangements". Curr. Protein Pept. Sci. 4 (3): 217–29. PMID 12769720. doi:10.2174/1389203033487199.
- ↑ Leonardi R, Zhang YM, Rock CO, Jackowski S (2005). "Coenzyme A: back in action". Prog. Lipid Res. 44 (2–3): 125–53. PMID 15893380. doi:10.1016/j.plipres.2005.04.001.
- ↑ Donnelly JG (2001). "Folic acid". Crit Rev Clin Lab Sci. 38 (3): 183–223. PMID 11451208. doi:10.1080/20014091084209.
- ↑ Søballe B, Poole RK (1999). "Microbial ubiquinones: multiple roles in respiration, gene regulation and oxidative stress management" (PDF). Microbiology (Reading, Engl.). 145 (8): 1817–30. PMID 10463148. doi:10.1099/13500872-145-8-1817.
- ↑ Linster CL, Van Schaftingen E (2007). "Vitamin C. Biosynthesis, recycling and degradation in mammals". FEBS J. 274 (1): 1–22. PMID 17222174. doi:10.1111/j.1742-4658.2006.05607.x.
- 1 2 Joosten V, van Berkel WJ (2007). "Flavoenzymes". Curr Opin Chem Biol. 11 (2): 195–202. PMID 17275397. doi:10.1016/j.cbpa.2007.01.010.
- ↑ Mack M, Grill S (2006). "Riboflavin analogs and inhibitors of riboflavin biosynthesis". Appl. Microbiol. Biotechnol. 71 (3): 265–75. PMID 16607521. doi:10.1007/s00253-006-0421-7.
- ↑ Bugg, Tim (1997). An introduction to enzyme and coenzyme chemistry. Oxford: Blackwell Science. p. 95. ISBN 0-86542-793-3.
- ↑ Chiang P, Gordon R, Tal J, Zeng G, Doctor B, Pardhasaradhi K, McCann P (1996). "S-Adenosylmethionine and methylation". FASEB J. 10 (4): 471–80. PMID 8647346.
- ↑ Noll KM, Rinehart KL, Tanner RS, Wolfe RS (1986). "Structure of component B (7-mercaptoheptanoylthreonine phosphate) of the methylcoenzyme M methylreductase system of Methanobacterium thermoautotrophicum". Proc. Natl. Acad. Sci. U.S.A. 83 (12): 4238–42. PMC 323707 . PMID 3086878. doi:10.1073/pnas.83.12.4238.
- ↑ Taylor CD, Wolfe RS (10 August 1974). "Structure and methylation of coenzyme M(HSCH2CH2SO3)". J. Biol. Chem. 249 (15): 4879–85. PMID 4367810.
- ↑ Balch WE, Wolfe RS (1979). "Specificity and biological distribution of coenzyme M (2-mercaptoethanesulfonic acid)". J. Bacteriol. 137 (1): 256–63. PMC 218444 . PMID 104960.
- ↑ Crane FL (1 December 2001). "Biochemical functions of coenzyme Q10". Journal of the American College of Nutrition. 20 (6): 591–8. PMID 11771674. doi:10.1080/07315724.2001.10719063. Archived from the original on 16 December 2008.
- ↑ Buchanan; Gruissem, Jones (2000). Biochemistry & molecular biology of plants (1st ed.). American society of plant physiology. ISBN 0-943088-39-9.
- ↑ Grill D, Tausz T, De Kok LJ (2001). Significance of glutathione in plant adaptation to the environment. Springer. ISBN 1-4020-0178-9.
- ↑ Meister A, Anderson ME (1983). "Glutathione". Annu. Rev. Biochem. 52: 711–60. PMID 6137189. doi:10.1146/annurev.bi.52.070183.003431.
- ↑ Wijayanti N, Katz N, Immenschuh S (2004). "Biology of heme in health and disease". Curr. Med. Chem. 11 (8): 981–6. PMID 15078160. doi:10.2174/0929867043455521.
- ↑ Vorholt JA, Thauer RK (1997). "The active species of 'CO2' utilized by formylmethanofuran dehydrogenase from methanogenic Archaea". Eur. J. Biochem. 248 (3): 919–24. PMID 9342247. doi:10.1111/j.1432-1033.1997.00919.x.
- ↑ Mendel RR, Hänsch R (2002). "Molybdoenzymes and molybdenum cofactor in plants". J. Exp. Bot. 53 (375): 1689–98. PMID 12147719. doi:10.1093/jxb/erf038.
- ↑ Mendel RR, Bittner F (2006). "Cell biology of molybdenum". Biochim. Biophys. Acta. 1763 (7): 621–35. PMID 16784786. doi:10.1016/j.bbamcr.2006.03.013.
- ↑ Ginsburg V (1978). "Comparative biochemistry of nucleotide-linked sugars". Prog. Clin. Biol. Res. 23: 595–600. PMID 351635.
- ↑ Negishi M, Pedersen LG, Petrotchenko E, et al. (2001). "Structure and function of sulfotransferases". Arch. Biochem. Biophys. 390 (2): 149–57. PMID 11396917. doi:10.1006/abbi.2001.2368.
- ↑ Salisbury SA, Forrest HS, Cruse WB, Kennard O (August 1979). "A novel coenzyme from bacterial primary alcohol dehydrogenases". Nature. 280 (5725): 843–4. PMID 471057. doi:10.1038/280843a0.
- ↑ Thony B, Auerbach G, Blau N (2000). "Tetrahydrobiopterin biosynthesis, regeneration and functions". Biochem J. 347 (1): 1–16. PMC 1220924 . PMID 10727395. doi:10.1042/0264-6021:3470001.
- ↑ DiMarco AA, Bobik TA, Wolfe RS (1990). "Unusual coenzymes of methanogenesis". Annu. Rev. Biochem. 59: 355–94. PMID 2115763. doi:10.1146/annurev.bi.59.070190.002035.
- ↑ Mitchell P (1979). "The Ninth Sir Hans Krebs Lecture. Compartmentation and communication in living systems. Ligand conduction: a general catalytic principle in chemical, osmotic and chemiosmotic reaction systems". Eur J Biochem. 95 (1): 1–20. PMID 378655. doi:10.1111/j.1432-1033.1979.tb12934.x.
- ↑ Wimmer M, Rose I (1978). "Mechanisms of enzyme-catalyzed group transfer reactions". Annu Rev Biochem. 47: 1031–78. PMID 354490. doi:10.1146/annurev.bi.47.070178.005123.
- ↑ Di Carlo SE, Collins HL (2001). "Estimating ATP resynthesis during a marathon run: a method to introduce metabolism". Advan. Physiol. Edu. 25 (2): 70–1.
- ↑ Chen X, Li N, Ellington AD (2007). "Ribozyme catalysis of metabolism in the RNA world". Chem. Biodivers. 4 (4): 633–55. PMID 17443876. doi:10.1002/cbdv.200790055.
- ↑ Koch A (1998). "How did bacteria come to be?". Adv Microb Physiol. 40: 353–99. PMID 9889982. doi:10.1016/S0065-2911(08)60135-6.
- ↑ Ouzounis C, Kyrpides N (1996). "The emergence of major cellular processes in evolution". FEBS Lett. 390 (2): 119–23. PMID 8706840. doi:10.1016/0014-5793(96)00631-X.
- ↑ White HB (1976). "Coenzymes as fossils of an earlier metabolic state". J. Mol. Evol. 7 (2): 101–4. PMID 1263263. doi:10.1007/BF01732468.
- ↑ Saran D, Frank J, Burke DH (2003). "The tyranny of adenosine recognition among RNA aptamers to coenzyme A". BMC Evol. Biol. 3: 26. PMC 317284 . PMID 14687414. doi:10.1186/1471-2148-3-26.
- ↑ Jadhav VR, Yarus M (2002). "Coenzymes as coribozymes". Biochimie. 84 (9): 877–88. PMID 12458080. doi:10.1016/S0300-9084(02)01404-9.
- ↑ Khoury, GA; Fazelinia, H; Chin, JW; Pantazes, RJ; Cirino, PC; Maranas, CD (October 2009). "Computational design of Candida boidinii xylose reductase for altered cofactor specificity". Protein Science. 18 (10): 2125–38. PMC 2786976 . PMID 19693930. doi:10.1002/pro.227
- ↑ Harden A, Young WJ (24 October 1906). "The Alcoholic Ferment of Yeast-Juice" (PDF). Proceedings of the Royal Society B: Biological Sciences. 78 (526): 369–75. doi:10.1098/rspb.1906.0070.
- ↑ "Fermentation of sugars and fermentative enzymes: Nobel Lecture, May 23, 1930" (PDF). Nobel Foundation. Retrieved 2007-09-30.
- ↑ Lohmann K (August 1929). "Über die Pyrophosphatfraktion im Muskel". Naturwissenschaften. 17 (31): 624–5. doi:10.1007/BF01506215.
- ↑ Lipmann F (1 September 1945). "Acetylation of sulfanilamide by liver homogenates and extracts". J. Biol. Chem. 160 (1): 173–90.
- ↑ Warburg O, Christian W (1936). "Pyridin, the hydrogen-transferring component of the fermentation enzymes (pyridine nucleotide)". Biochemische Zeitschrift. 287: 291. doi:10.1002/hlca.193601901199.
- ↑ Kalckar HM (1974). "Origins of the concept oxidative phosphorylation". Mol. Cell. Biochem. 5 (1–2): 55–63. PMID 4279328. doi:10.1007/BF01874172.
- ↑ Lipmann F (1941). "Metabolic generation and utilization of phosphate bond energy". Adv Enzymol. 1: 99–162. doi:10.4159/harvard.9780674366701.c141.
- ↑ Friedkin M, Lehninger AL (1949). "Esterification of inorganic phosphate coupled to electron transport between dihydrodiphosphopyridine nucleotide and oxygen". J. Biol. Chem. 178 (2): 611–23. PMID 18116985.
- 1 2 Davidson, Victor L. "Protein-Derived Cofactors. Expanding the Scope of Post-Translational Modifications†". Biochemistry. 46 (18): 5283–5292. doi:10.1021/bi700468t.
- ↑ Davidson VL, Wilmot CM (2013). "Posttranslational biosynthesis of the protein-derived cofactor tryptophan tryptophylquinone.". Annu. Rev. Biochem. 82: 531–50. PMC 4082410 . PMID 23746262. doi:10.1146/annurev-biochem-051110-133601.
- ↑ Huang SX, Lohman JR, Huang T, Shen B (May 2013). "A new member of the 4-methylideneimidazole-5-one-containing aminomutase family from the enediyne kedarcidin biosynthetic pathway.". Proc. Natl. Acad. Sci. U.S.A. 110 (20): 8069–74. PMC 3657804 . PMID 23633564. doi:10.1073/pnas.1304733110.
- ↑ Lodish, Harvey; Berk, Arnold; Zipursky, S. Lawrence; Matsudaira, Paul; Baltimore, David; Darnell, James (2000-01-01). "G Protein –Coupled Receptors and Their Effectors".
- ↑ McKenna NJ, O'Malley BW (October 2008). "Editorial: Coactivators and Corepressors: What’s in a Name?.". Mol. Endocrinol. 22 (10): 2213–4. PMC 2582534 . PMID 18701638. doi:10.1210/me.2008-0201.
Further reading
- Bugg, Tim (1997). An introduction to enzyme and coenzyme chemistry. Oxford: Blackwell Science. ISBN 0-86542-793-3.
External links
- Cofactors lecture (Powerpoint file)
- Enzyme cofactors at the US National Library of Medicine Medical Subject Headings (MeSH)
- The CoFactor Database