Chimeric antigen receptor
Chimeric antigen receptors (CARs), (also known as chimeric immunoreceptors, chimeric T cell receptors, artificial T cell receptors) are engineered receptors, which graft an arbitrary specificity onto an immune effector cell (T cell). Typically, these receptors are used to graft the specificity of a monoclonal antibody onto a T cell, with transfer of their coding sequence facilitated by retroviral vectors. The receptors are called chimeric because they are composed of parts from different sources.
CARs are under investigation as a therapy for cancer, using a technique called adoptive cell transfer.[1] T cells are removed from a patient and modified so that they express receptors specific to the patient's particular cancer. The T cells, which can then recognize and kill the cancer cells, are reintroduced into the patient. Modification of T-cells sourced from donors other than the patient are also under investigation.
Structure
The most common form of CARs are fusions of single-chain variable fragments (scFv) derived from monoclonal antibodies, fused to CD3-zeta transmembrane- and endodomain. An example of such a construct is 14g2a-Zeta, which is a fusion of a scFv derived from hybridoma 14g2a (which recognizes disialoganglioside GD2).
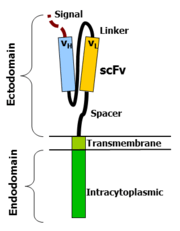
The variable portions of an immunoglobulin heavy and light chain are fused by a flexible linker to form an scFv. This scFv is preceded by a signal peptide to direct the nascent protein to the endoplasmic reticulum and subsequent surface expression (cloven). A flexible spacer allows the scFv to orient in different directions to enable antigen binding. The transmembrane domain is a typical hydrophobic alpha helix usually derived from the original molecule of the signalling endodomain that protrudes into the cell and transmits the desired signal.
Type I proteins are in fact two protein domains linked by a transmembrane alpha helix. The cell membrane lipid bilayer, through which the transmembrane domain passes, isolates the inside portion (endodomain) from the external portion (ectodomain). Attaching an ectodomain from one protein to an endodomain of another protein makes a molecule that combines the recognition of the former to the signal of the latter.
ScFv/CD3-zeta hybrids result in the transmission of a zeta signal in response to recognition by the scFv of its target. When T cells express this molecule (usually achieved by oncoretroviral vector transduction), they recognize and kill target cells that express GD2 (e.g. neuroblastoma cells). To target malignant B cells, investigators have redirected the specificity of T cells using a chimeric immunoreceptor specific for the B-lineage molecule, CD19.
Ectodomain
The ectodomain is the domain of a membrane protein which is outside the cell, outside the cytoplasm [extracytoplasmic] and exposed to the extracellular space (when not inside the lumen on intracellular vesicles in the secretory or endocytic pathways). For a cell surface receptor or protein, this will be the domain exposed to the extracellular space.
Signal peptide
A signal peptide directs the nascent protein into the endoplasmic reticulum. This is essential if the receptor is to glycosylate and anchor in the cell membrane. Any eukaryotic signal peptide sequence usually works. Generally, the signal peptide natively attached to the amino-terminal most component is used (e.g. in a scFv with orientation light chain - linker - heavy chain, the native signal of the light-chain is used).
Antigen recognition region
The antigen recognition region is usually a scFv, although many alternatives exist. An antigen recognition domain from native T-cell receptor (TCR) alpha and beta single chains have been described, as have simple ectodomains (e.g. CD4 ectodomain to recognize HIV infected cells) and more exotic recognition components such as a linked cytokine (which leads to recognition of cells bearing the cytokine receptor). Almost anything that binds a given target with high affinity can be used as an antigen recognition region.
Spacer
A spacer region links the antigen binding domain to the transmembrane domain. It should be flexible enough to allow the antigen binding domain to orient in different directions to facilitate antigen recognition. The simplest form is the hinge region from IgG1. Alternatives include the CH2CH3 region of immunoglobulin and portions of CD3. For most scFv based constructs, the IgG1 hinge suffices. However the best spacer often is determined empirically.
Transmembrane domain
The transmembrane domain is a hydrophobic alpha helix that spans the membrane. Generally, the transmembrane domain from the most membrane proximal component of the endodomain is used. Interestingly, using the CD3-zeta transmembrane domain may result in incorporation of the artificial TCR into the native TCR, a factor that is dependent on the presence of the native CD3-zeta transmembrane charged aspartic acid residue.[2] Different transmembrane domains result in different receptor stability. The CD28 transmembrane domain results in a brightly expressed, stable receptor.
Endodomain
This is the functional end of the receptor. After antigen recognition, receptors cluster and a signal is transmitted to the cell. The most commonly used endodomain component is CD3-zeta which contains 3 ITAMs. This transmits an activation signal to the T cell after the antigen is bound. CD3-zeta may not provide a fully competent activation signal and co-stimulatory signaling is needed. For example, chimeric CD28 and OX40 can be used with CD3-Zeta to transmit a proliferative/survival signal or all three can be used together.
Evolution of CAR T-cell design

First generation CARs typically had the intracellular domain from the CD3 ζ- chain, which is the primary transmitter of signals from endogenous TCRs. Second generation CARs add intracellular signaling domains from various costimulatory protein receptors (e.g., CD28, 41BB, ICOS) to the cytoplasmic tail of the CAR to provide additional signals to the T cell. Preclinical studies indicated that the second generation improves the antitumor activity of T cells. More recent, third generation CARs combine multiple signaling domains, such as CD3z-CD28-41BB or CD3z-CD28-OX40, to augment potency.
The evolution of CAR therapy is an excellent example of the application of basic research to the clinic. The PI3K binding site used was identified in co-receptor CD28,[4] while the ITAM motifs were identified as a target of the CD4- and CD8-p56lck complexes.[5]
The introduction of Strep-tag II sequence (an eight-residue minimal peptide sequence (Trp-Ser-His-Pro-Gln-Phe-Glu-Lys) that exhibits intrinsic affinity toward streptavidin[6]) into specific sites in synthetic CARs or natural T-cell receptors provides engineered T cells with an identification marker for rapid purification, a method for tailoring spacer length of chimeric receptors for optimal function and a functional element for selective antibody-coated, microbead-driven, large-scale expansion.[7][8] Strep-tag can be used to stimulate the engineered cells, causing them to grow rapidly. Using an antibody that binds the Strep-tag, the engineered cells can be expanded by 200-fold. Unlike existing methods this technology stimulates only cancer-specific T cells.
SMDC adaptor technology
SMDCs (small molecule drug conjugates) platform in immuno-oncology is a novel (currently experimental) approach that makes possible the engineering of a single universal CAR T cell, which binds with extraordinarily high affinity to a benign molecule designated as FITC. These cells are then used to treat various cancer types when co-administered with bispecific SMDC adaptor molecules. These unique bispecific adaptors are constructed with a FITC molecule and a tumor-homing molecule to precisely bridge the universal CAR T cell with the cancer cells, which causes localized T cell activation. Anti-tumor activity in mice is induced only when both the universal CAR T cells plus the correct antigen-specific adaptor molecules are present. Anti-tumor activity and toxicity can be controlled by adjusting the administered adaptor molecule dosing. Treatment of antigenically heterogeneous tumors can be achieved by administration of a mixture of the desired antigen-specific adaptors. Thus, several challenges of current CAR T cell therapies, such as:
- the inability to control the rate of cytokine release and tumor lysis
- the absence of an “off switch” that can terminate cytotoxic activity when tumor eradication is complete
- a requirement to generate a different CAR T cell for each unique tumor antigen
may be solved or mitigated using this approach.[9][10][11]
History
The concept of doctoring T-cells genetically was developed in the 1980s by Eshhar and colleagues. By 1989, Eshhar and his colleagues had created the first functional CAR T cells.[12]
Clinical studies
The use of CARs in the clinic is based on reprogramming the T cell antigen receptor using a vector (for example viral) that is specific for malignant cells. Pre-clinical and clinical trials have focused on optimizing structure and signalling.
The first generation of CAR-modified T cells (CARTs) showed success in pre-clinical trials and have entered phase I clinical trials in ovarian cancer, neuroblastoma and various types of leukemia and lymphoma. To date, these clinical trials have shown little evidence of anti-tumor activity, with insufficient activation, persistence and homing to cancer tissue. Some anti-tumor responses have been reported in patients with B cell lymphoma (treated with alfaCD20-CD3zeta CAR-modified T cells) and some neuroblastoma patients (treated with ScFv-CD3zeta CARTs) reported partial response, stable disease and remission.
Second and third generation CARTs provided enhanced activation signals, proliferation, production of cytokines and effector function in pre-clinical trials. Both second and the third generation CARTs are entering clinical trials. In a study with alfaCD19.4-1BB.CD3zeta CARTs in patients with chronic lymphocyte leukemia, complete remission was ongoing 10 months after treatment. CARTs cells were found to expand 3-logs in these patients, and to have infiltrated and lysed cancer tissue. A fraction of these cells displayed a memory phenotype for preventive tumor relapses. Although these CARTs produced significant therapeutic effect, their activity led to life-threatening tumorlysis 3 weeks after the first infusion.
Adverse events were reported that highlight the need for caution while using second and third generation CARTs. One patient died 5 days after cyclophosphamide chemotherapy followed by infusion of CARTs recognizing the antigen ERBB2 (HER-2/neu).[13] The toxicity led to a clinically significant release of pro-inflammatory cytokines, pulmonary toxicity, multi-organ failure and eventual patient death. This "cytokine storm" (cytokine release syndrome) was thought to be due to CAR T cell cytotoxicity against normal lung epithelial cells, known to express low levels of ERBB2. This and other adverse events highlight the need for caution when employing CARTs, as unlike antibodies against tumor-associated antigens, these cells are not cleared from the body quickly. Long exposure to CARTs is necessary for good clinical outcome, but is not feasible due to adverse effects, particularly prolonged absence of myelopoiesis.[14] Efforts to circumnavigate this problem with a suicide gene are currently underway.[14]
A 2016 review indicates that recent clinical trials evaluating active immunization or immunoconjugates to mesothelin in patients with pancreatic adenocarcinoma or mesothelioma have shown responses without toxicity.[15]
The great promise of cancer immunotherapy is to clear the tumor without the toxicity of conventional treatments. The treatment of cancer with CARTs has several advantages: HLA-independent recognition of antigen, broad applicability for many patients and rapid delivery. Successful application of CARTs will require the identification of a tumor-associated antigen that is expressed only on tumor cells, thereby minimizing toxicity risk,[16][17]
Early examples
A list of tumors antigens and CARs in in vitro and in vivo trials As of 2012:[17][18]
| Target antigen | Associated malignancy | Receptor type | CARs generation |
|---|---|---|---|
| α-Folate receptor | Ovarian cancer | ScFv-FcεRIγCAIX | First |
| CAIX | Renal cell carcinoma | ScFv-FcεRIγ | First |
| CAIX | Renal cell carcinoma | ScFv-FcεRIγ | Second |
| CD19 | B-cell malignancies | ScFv-CD3ζ (EBV) | First |
| CD19 | B-cell malignancies, CLL | ScFv-CD3ζ | First |
| CD19 | B-ALL | ScFv-CD28-CD3ζ | Second |
| CD19 | ALL | CD3ζ(EBV) | First |
| CD19 | ALL post-HSCT | ScFv-CD28-CD3ζ | Second |
| CD19 | Leukemia, lymphoma, CLL | ScFv-CD28-CD3ζ vs. CD3ζ | First and Second |
| CD19 | B-cell malignancies | ScFv-CD28-CD3ζ | Second |
| CD19 | B-cell malignancies post-HSCT | ScFv-CD28-CD3ζ | Second |
| CD19 | Refractory Follicular Lymphoma | ScFv-CD3ζ | First |
| CD19 | B-NHL | ScFv -CD3ζ | First |
| CD19 | B-lineage lymphoid malignancies post-UCBT | ScFv-CD28-CD3ζ | Second |
| CD19 | CLL, B-NHL | ScFv-CD28-CD3ζ | Second |
| CD19 | B-cell malignancies, CLL, B-NHL | ScFv-CD28-CD3ζ | Second |
| CD19 | ALL, lymphoma | ScFv-41BB-CD3ζ vs CD3ζ | First and Second |
| CD19 | ALL | ScFv-41BB-CD3ζ | Second |
| CD19 | B-cell malignancies | ScFv-CD3ζ (Influenza MP-1) | First |
| CD19 | B-cell malignancies | ScFv-CD3ζ (VZV) | First |
| CD20 | Lymphomas | ScFv-CD28-CD3ζ | Second |
| CD20 | B-cell malignancies | ScFv-CD4-CD3ζ | Second |
| CD20 | B-cell lymphomas | ScFv-CD3ζ | First |
| CD20 | Mantle cell lymphoma | ScFv-CD3ζ | First |
| CD20 | Mantle cell lymphoma, indolent B-NHL | CD3 ζ /CD137/CD28 | Third |
| CD20 | indolent B cell lymphomas | ScFv-CD28-CD3ζ | Second |
| CD20 | Indolent B cell lymphomas | ScFv-CD28-41BB-CD3ζ | Third |
| CD22 | B-cell malignancies | ScFV-CD4-CD3ζ | Second |
| CD30 | Lymphomas | ScFv-FcεRIγ | First |
| CD30 | Hodgkin lymphoma | ScFv-CD3ζ (EBV) | First |
| CD33 | AML | ScFv-CD28-CD3ζ | Second |
| CD33 | AML | ScFv-41BB-CD3ζ | Second |
| CD44v7/8 | Cervical carcinoma | ScFv-CD8-CD3ζ | Second |
| CEA | Breast cancer | ScFv-CD28-CD3ζ | Second |
| CEA | Colorectal cancer | ScFv-CD3ζ | First |
| CEA | Colorectal cancer | ScFv-FceRIγ | First |
| CEA | Colorectal cancer | ScFv-CD3ζ | First |
| CEA | Colorectal cancer | ScFv-CD28-CD3ζ | Second |
| CEA | Colorectal cancer | ScFv-CD28-CD3ζ | Second |
| EGP-2 | Multiple malignancies | scFv-CD3ζ | First |
| EGP-2 | Multiple malignancies | scFv-FcεRIγ | First |
| EGP-40 | Colorectal cancer | scFv-FcεRIγ | First |
| erb-B2 | Colorectal cancer | CD28/4-1BB-CD3ζ | Third |
| erb-B2 | Breast and others | ScFv-CD28-CD3ζ | Second |
| erb-B2 | Breast and others | ScFv-CD28-CD3ζ (Influenza) | Second |
| erb-B2 | Breast and others | ScFv-CD28mut-CD3ζ | Second |
| erb-B2 | Prostate cancer | ScFv-FcεRIγ | First |
| erb-B 2,3,4 | Breast and others | Heregulin-CD3ζ | Second |
| erb-B 2,3,4 | Breast and others | ScFv-CD3ζ | First |
| FBP | Ovarian cancer | ScFv-FcεRIγ | First |
| FBP | Ovarian cancer | ScFv-FcεRIγ (alloantigen) | First |
| Fetal acetylcholine receptor | Rhabdomyosarcoma | ScFv-CD3ζ | First |
| GD2 | Neuroblastoma | ScFv-CD28 | First |
| GD2 | Neuroblastoma | ScFv-CD3ζ | First |
| GD2 | Neuroblastoma | ScFv-CD3ζ | First |
| GD2 | Neuroblastoma | ScFv-CD28-OX40-CD3ζ | Third |
| GD2 | Neuroblastoma | ScFv-CD3ζ (VZV) | First |
| GD3 | Melanoma | ScFv-CD3ξ | First |
| GD3 | Melanoma | ScFv-CD3ξ | First |
| Her2/neu | Medulloblastoma | ScFv-CD3ξ | First |
| Her2/neu | Lung malignancy | ScFv-CD28-CD3ζ | Second |
| Her2/neu | Advanced osteosarcoma | ScFv-CD28-CD3ζ | Second |
| Her2/neu | Glioblastoma | ScFv-CD28-CD3ζ | Second |
| IL-13R-a2 | Glioma | IL-13-CD28-4-1BB-CD3ζ | Third |
| IL-13R-a2 | Glioblastoma | IL-13-CD3ζ | Second |
| IL-13R-a2 | Medulloblastoma | IL-13-CD3ζ | Second |
| KDR | Tumor neovasculature | ScFv-FcεRIγ | First |
| k-light chain | B-cell malignancies | ScFv-CD3ζ | First |
| k-light chain | (B-NHL, CLL) | ScFv-CD28-CD3ζ vs CD3ζ | Second |
| LeY | Carcinomas | ScFv-FcεRIγ | First |
| LeY | Epithelial derived tumors | ScFv-CD28-CD3ζ | Second |
| L1 cell adhesion molecule | Neuroblastoma | ScFv-CD3ζ | First |
| MAGE-A1 | Melanoma | ScFV-CD4-FcεRIγ | Second |
| MAGE-A1 | Melanoma | ScFV-CD28-FcεRIγ | Second |
| Mesothelin | Various tumors | ScFv-CD28-CD3ζ | Second |
| Mesothelin | Various tumors | ScFv-41BB-CD3ζ | Second |
| Mesothelin | Various tumors | ScFv-CD28-41BB-CD3ζ | Third |
| Murine CMV infected cells | Murine CMV | Ly49H-CD3ζ | Second |
| MUC1 | Breast, Ovary | ScFV-CD28-OX40-CD3ζ | Third |
| NKG2D ligands | Various tumors | NKG2D-CD3ζ | First |
| Oncofetal antigen (h5T4) | Various tumors | ScFV-CD3ζ (vaccination) | First |
| PSCA | Prostate carcinoma | ScFv-b2c-CD3ζ | Second |
| PSMA | Prostate/tumor vasculature | ScFv-CD3ζ | First |
| PSMA | Prostate/tumor vasculature | ScFv-CD28-CD3ζ | Second |
| PSMA | Prostate/tumor vasculature | ScFv-CD3ζ | First |
| TAA targeted by mAb IgE | Various tumors | FceRI-CD28-CD3ζ (+ a-TAA IgE mAb) | Third |
| TAG-72 | Adenocarcinomas | scFv-CD3ζ | First |
| VEGF-R2 | Tumor neovasculature | scFv-CD3ζ | First |
Adoptive transfer of CAR-modified cells as a cancer therapeutic
Adoptive transfer of T cells expressing chimeric antigen receptors is a promising anti-cancer therapeutic as CAR-modified T cells can be engineered to target virtually any tumor associated antigen. There is great potential for this approach to improve patient-specific cancer therapy in a profound way. Following the collection of a patient's T cells, the cells are genetically engineered to express CARs specifically directed towards antigens on the patient's tumor cells, then infused back into the patient.[19]
Engineering CAR T cells
A common method for engineering CAR T cells for cancer immunotherapy is to use viral vectors such as retrovirus, lentivirus or transposon, that integrate the transgene in the host cell genome. However, this poses a risk of negatively affecting the T cell’s endogenous gene expression and has raised concerns of genotoxicity in the engineered T cells, i.e., that they can become tumorigenic. Alternatively, non-integrating vectors such as plasmids or mRNA can be used but these types of episomal DNA/RNA are lost upon repeated cell division and the engineered CAR T cells will eventually lose their CAR expression after about 1 week.[20][21] There is also a third possibility, where a vector is used that is stably maintained in the T cell, without being integrated in its genome. This strategy has been found to enable long-term transgene expression without the risk of insertional mutagenesis or genotoxicity, making it feasible as a safe approach to produce CAR T cells for cancer immunotherapy.[22]
Safety concerns
Although adoptive transfer of CAR-modified T-cells is a unique and promising cancer therapeutic, there are significant safety concerns. Clinical trials of this therapy have revealed potential toxic effects of these CARs when healthy tissues express the same target antigens as the tumor cells, leading to outcomes similar to graft-versus-host disease (GVHD). A potential solution to this problem is engineering a suicide gene into the modified T cells. In this way, administration of a prodrug designed to activate the suicide gene during GVHD triggers apoptosis in the suicide gene-activated CAR T cells. This method has been used safely and effectively in hematopoietic stem cell transplantation (HSCT). Adoption of suicide gene therapy to the clinical application of CAR-modified T cell adoptive cell transfer has potential to alleviate GVHD while improving overall anti-tumor efficacy.[3]
Differences between T-Cell Receptor therapy and Chimeric Antigen Receptor T-cell therapy
1. Link https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4381333/
2. Link http://www.nature.com/nature/journal/v504/n7480_supp/full/504S13a.html
See also
References
- ↑ Pule, M; Finney H; Lawson A (2003). "Artificial T-cell receptors". Cytotherapy. 5 (3): 211–26. PMID 12850789. doi:10.1080/14653240310001488.
- ↑ Bridgeman, JS; Hawkins RE; Bagley S; Blaylock M; Holland M; Gilham DE (2010). "The Optimal Antigen Response of Chimeric Antigen Receptors Harbouring the CD3zeta Transmembrane Domain Is Dependent Upon Incorporation of the Receptor Into the Endogenous TCR/CD3 Complex". Journal of Immunology. 184 (12): 6938–49. PMID 20483753. doi:10.4049/jimmunol.0901766.
- 1 2 Casucci, Monica; Attilio Bondanza (2011). "Suicide Gene Therapy to Increase the Safety of Chimeric Antigen Receptor-Redirected T Lymphocytes". Journal of Cancer. 2: 378–382. PMC 3133962
 . PMID 21750689. doi:10.7150/jca.2.378. Retrieved 30 April 2012.
. PMID 21750689. doi:10.7150/jca.2.378. Retrieved 30 April 2012. - ↑ Rudd, Christopher E.; Schneider, Helga (July 2003). "Unifying concepts in CD28, ICOS and CTLA4 co-receptor signalling". Nature Reviews Immunology. 3 (7): 544–556. PMID 12876557. doi:10.1038/nri1131.
- ↑ Rudd, Christopher E. (1999-01-08). "Adaptors and Molecular Scaffolds in Immune Cell Signaling". Cell. 96 (1): 5–8. ISSN 0092-8674. doi:10.1016/S0092-8674(00)80953-8.
- ↑ Schmidt, T. G., & Skerra, A. (2007). The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nature Protocols, 2(6), 1528-1535. doi:10.1038/nprot.2007.209
- ↑ Liu L., Sommermeyer D., Cabanov A., Kosasih P., Hill T. & Riddell S. R (2016). Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nature Biotechnology, doi:10.1038/nbt.3461
- ↑ Crafting a better T cell for immunotherapy. New technology aims to reduce patients’ waiting time, increase potency of T-cell therapy
- ↑ Research could expand engineered T-cell cancer treatment. Medical Xpress
- ↑ SMDC TECHNOLOGY. ENDOCYTE
- ↑ ENDOCYTE ANNOUNCES PROMISING PRECLINICAL DATA FOR APPLICATION OF SMDC TECHNOLOGY IN CAR T CELL THERAPY. Apr 19, 2016 ENDOCYTE
- ↑ Urba, Walter J.; Longo, Dan L. (2011-08-25). "Redirecting T Cells". New England Journal of Medicine. 365 (8): 754–757. ISSN 0028-4793. PMID 21830939. doi:10.1056/NEJMe1106965.
- ↑ Morgan, Richard; Yang, J. C.; Kitano, M.; Dudley, M. E.; Laurencot, C. M.; Rosenberg, S. A. (2010-02-25). "Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2". Molecular Therapy. 18 (4): 843–51. PMC 2862534 . PMID 20179677. doi:10.1038/mt.2010.24.
- 1 2 Gill, Saar (2016-12-01). "Chimeric antigen receptor T cell therapy in AML: How close are we?". Best Practice & Research Clinical Haematology. Acute Leukemia and Myelodysplasia: Advances and Controversies. 29 (4): 329–333. PMC 5131522 . PMID 27890255. doi:10.1016/j.beha.2016.10.004.
- ↑ Morello A.; et al. (February 2016). "Mesothelin-Targeted CARs: Driving T Cells to Solid Tumors.". Cancer Discovery. 6 (2): 133–46. doi:10.1158/2159-8290.CD-15-0583.
- ↑ Curran, Kevin J.; Pengram Hollie J; Brentjens Renier J (2012). "The promise and potential". J Gene Med. 14 (6): 405–415. PMID 22262649. doi:10.1002/jgm.2604. Retrieved 5 January 2012.
- 1 2 Lipowska-Bhalla, Grazyna; Gilham David E.; Hawkins Robert E.; Rothwell Dominic G. (2012). "Target immunotherapy of cancer with CAR T cell: achievements and challenges". Cancer Immunol Immunother. 61 (7): 953–962. PMID 22527245. doi:10.1007/s00262-012-1254-0. Retrieved 25 March 2012.
- ↑ Sadelain, M.; Gilham David E.; Hawkins Robert E.; Rothwell Dominic G. (2009). "The promise and potential pitfalls of chimeric antigen receptor". Curr Opin Immunol. 21 (2): 215–223. PMID 19327974. doi:10.1016/j.coi.2009.02.009.
- ↑ Jacobson, Caron; Jerome Ritz (3 November 2011). "Time to put the CAR-T before the horse". Blood. 118 (18): 4761–4762. PMID 22053170. doi:10.1182/blood-2011-09-376137. Retrieved 7 May 2012.
- ↑ Smits, E.; Ponsaerts, P.; Lenjou, M.; Nijs, G.; Van Bockstaele, D. R.; Berneman, Z. N.; Van Tendeloo, V. F. I. (2004-09-23). "RNA-based gene transfer for adult stem cells and T cells". Leukemia. 18 (11): 1898–1902. ISSN 0887-6924. doi:10.1038/sj.leu.2403463.
- ↑ Yoon, S. H.; Lee, J. M.; Cho, H. I.; Kim, E. K.; Kim, H. S.; Park, M. Y.; Kim, T. G. (2008-12-19). "Adoptive immunotherapy using human peripheral blood lymphocytes transferred with RNA encoding Her-2/neu-specific chimeric immune receptor in ovarian cancer xenograft model". Cancer Gene Therapy. 16 (6): 489–497. ISSN 0929-1903. doi:10.1038/cgt.2008.98.
- ↑ Jin, Chuan; Fotaki, Grammatiki; Ramachandran, Mohanraj; Nilsson, Berith; Essand, Magnus; Yu, Di (2016-07-01). "Safe engineering of CAR T cells for adoptive cell therapy of cancer using long‐term episomal gene transfer". EMBO Molecular Medicine. 8 (7): 702–711. ISSN 1757-4676. PMC 4931286 . PMID 27189167. doi:10.15252/emmm.201505869.