Catalytic triad

A catalytic triad refers to the three amino acid residues that function together at the centre of the active site of some hydrolase and transferase enzymes (e.g. proteases, amidases, esterases, acylases, lipases and β-lactamases). An Acid-Base-Nucleophile triad is a common motif for generating a nucleophilic residue for covalent catalysis.[1][2] The residues form a charge-relay network to polarise and activate the nucleophile, which attacks the substrate, forming a covalent intermediate which is then hydrolysed to regenerate free enzyme and release the product. The nucleophile is most commonly a serine or cysteine amino acid, but occasionally threonine. Because enzymes fold into complex three-dimensional structures, the residues of a catalytic triad can be far from each other along the amino-acid sequence (primary structure), however, they are brought close together in the final fold.[3]
As well as divergent evolution of function (and even the triad's nucleophile), catalytic triads show some of the best examples of convergent evolution. Chemical constraints on catalysis have led to the same catalytic solution independently evolving in at least 23 separate superfamilies.[2] Their mechanism of action is consequently one of the best studied in biochemistry.[4][5]
History
The enzymes trypsin and chymotrypsin were first purified in the 1930s.[6] A serine in each of trypsin and chymotrypsin was identified as the catalytic nucleophile (by diisopropyl fluorophosphate modification) in the 1950s.[7] The structure of chymotrypsin was solved by X-ray crystallography in the 1960s, showing the orientation of the catalytic triad in the active site.[8] Other proteases were sequenced and aligned to reveal a family of related proteases,[9][10][11] now called the S1 family. Simultaneously, the structures of the evolutionarily unrelated papain and subtilisin proteases were found to contain analogous triads. The 'charge-relay' mechanism for the activation of the nucleophile by the other triad members was proposed in the late 1960s.[12] As more protease structures were solved by X-ray crystallography in the 1970s and 80s, homologous (such as TEV protease) and analogous (such as papain) triads were found.[13][14][15] The MEROPS classification system in the 1990s and 2000s began classing proteases into structurally related enzyme superfamilies and so acts as a database of the convergent evolution of triads in over 20 superfamilies.[16][17] Understanding how chemical constraints on evolution led to the convergence of so many enzyme families on the same triad geometries has developed in the 2010s.[2] Of particular contention during the 1990s and early 2000s was the contribution of low-barrier hydrogen bonding to catalysis;[18][19] however, current thinking is that ordinary hydrogen bonding is sufficient to explain the mechanism.[20][21] The massive body of work on the charge-relay, covalent catalysis used by catalytic triads has led to the mechanism being the best characterised in all of biochemistry.[4][5]
Function
Enzymes that contain a catalytic triad use it for one of two reaction types: either to split a substrate (hydrolases) or to transfer one portion of a substrate over to a second substrate (transferases). Triads are an inter-dependent set of residues in the active site of an enzyme and act in concert with other residues (e.g. binding site and oxyanion hole) to achieve nucleophilic catalysis. These triad residues act together to make the nucleophile member highly reactive, generating a covalent intermediate with the substrate that is then resolved to complete catalysis.
Mechanism
Catalytic triads perform covalent catalysis using a residue as a nucleophile. The reactivity of the nucleophilic residue is increased by the functional groups of the other triad members. The nucleophile is polarised and oriented by the base, which is itself bound and stabilised by the acid.
Catalysis is performed in two stages. First, the activated nucleophile attacks the carbonyl carbon and forces the carbonyl oxygen to accept an electron, leading to a tetrahedral intermediate. The build-up of negative charge on this intermediate is typically stabilized by an oxanion hole within the active site. The intermediate then collapses back to a carbonyl, ejecting the first half of the substrate, but leaving the second half still covalently bound to the enzyme as an acyl-enzyme intermediate. The ejection of this first leaving group is often aided by donation of a proton by the base.
The second stage of catalysis is the resolution of the acyl-enzyme intermediate by the attack of a second substrate. If this substrate is water then the result is hydrolysis; if it is an organic molecule then the result is transfer of that molecule onto the first substrate. Attack by this second substrate forms a new tetrahedral intermediate, which resolves by ejecting the enzyme's nucleophile, releasing the second product and regenerating free enzyme.

Identity of triad members
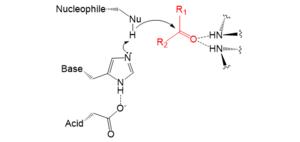
Nucleophile
The side-chain of the nucleophilic residue performs covalent catalysis on the substrate. The lone pair of electrons present on the oxygen or sulphur attacks the electropositive carbonyl carbon.[3] The 20 naturally occurring biological amino acids do not contain any sufficiently nucleophilic functional groups for many difficult catalytic reactions. Embedding the nucleophile in a triad increases its reactivity for efficient catalysis. The most commonly used nucleophiles are the hydroxyl (OH) of serine and the thiol/thiolate ion (SH/S−) of cysteine.[2] Alternatively, threonine proteases use the secondary hydroxyl of threonine, however due to steric hindrance of the side chain's extra methyl group such proteases use their N-terminal amide as the base, rather than a separate amino acid.[1][22]
Use of oxygen or sulphur as the nucleophilic atom causes minor differences in catalysis. Compared to oxygen, sulphur’s extra d orbital makes it larger (by 0.4 Å)[23] and softer, allows it to form longer bonds (dC-X and dX-H by 1.3-fold), and gives it a lower pKa (by 5 units).[24] Serine is therefore more dependent than cysteine on optimal orientation of the acid-base triad members to reduce its pKa[24] in order to achieve concerted deprotonation with catalysis.[2] The low pKa of cysteine works to its disadvantage in the resolution of the first tetrahedral intermediate as unproductive reversal of the original nucleophilic attack is the more favourable breakdown product.[2] The triad base is therefore preferentially oriented to protonate the leaving group amide to ensure that it is ejected to leave the enzyme sulphur covalently bound to the substrate N-terminus. Finally, resolution of the acyl-enzyme (to release the substrate C-terminus) requires serine to be re-protonated whereas cysteine can leave as S−. Sterically, the sulphur of cysteine also forms longer bonds and has a bulkier van der Waals radius[2] and if mutated to serine can be trapped in unproductive orientations in the active site.[23]
Base
Since no natural amino acids are strongly nucleophilic, the base in a catalytic triad polarises and deprotonates the nucleophile to increase its reactivity.[3] Additionally, it protonates the first product to aid leaving group departure.
The base is most commonly histidine since its pKa allows for effective base catalysis, hydrogen bonding to the acid residue, and deprotonation of the nucleophile residue.[1] β-lactamases such as TEM-1 use a lysine residue as the base. Because lysine's pKa is so high (pKa=11), a glutamate and several other residues act as the acid to stabilise its deprotonated state during the catalytic cycle.[25][26] Threonine proteases use their N-terminal amide as the base, since steric crowding by the catalytic threonine's methyl prevents other residues from being close enough.[27][28]
Acid
The acidic triad member forms a hydrogen bond with the basic residue. This aligns the basic residue by restricting its side-chain rotation, and polarises it by stabilising its positive charge.[3] Two amino acids have acidic side chains at physiological pH (aspartate or glutamate) and so are the most commonly used for this triad member.[3] Cytomegalovirus protease[lower-alpha 2] uses a pair of histidines, one as the base, as usual, and one as the acid.[1] The second histidine is not as effective an acid as the more common aspartate or glutamate, leading to a lower catalytic efficiency. In some enzymes, the acid member of the triad is less necessary and some act only as a dyad. For example, papain[lower-alpha 3] uses asparagine as its third triad member which orients the histidine base but does not act as an acid. Similarly, hepatitis A virus protease[lower-alpha 4] contains an ordered water in the position where an acid residue should be.
Examples of triads

Ser-His-Asp
The Serine-Histidine-Aspartate motif is one of the most thoroughly characterised catalytic motifs in biochemistry.[3] The triad is exemplified by chymotrypsin,[lower-alpha 5] a model serine protease from the PA superfamily which uses its triad to hydrolyse protein backbones. The aspartate is hydrogen bonded to the histidine, increasing the pKa of its imidazole nitrogen from 7 to around 12. This allows the histidine to act as a powerful general base and to activate the serine nucleophile. It also has an oxyanion hole consisting of several backbone amides which stabilises charge build-up on intermediates. The histidine base aids the first leaving group by donating a proton, and also activates the hydrolytic water substrate by abstracting a proton as the remaining OH− attacks the acyl-enzyme intermediate.
The same triad has also convergently evolved in α/β hydrolases such as some lipases and esterases, however orientation of the triad members is reversed. Additionally, brain acetyl hydrolase (which has the same fold as a small G-protein) has also been found to have this triad. The equivalent Serine-Histidine-Glutamate triad is used in acetylcholinesterase.
Cys-His-Asp
The second most studied triad is the Cysteine-Histidine-Aspartate motif.[2] Several families of cysteine proteases use this triad set, for example TEV protease[lower-alpha 1] and papain.[lower-alpha 3] The triad acts similarly to serine protease triads, with a few notable differences. Due to cysteine's low pKa, the importance of the Asp to catalysis varies and several cysteine proteases are effectively Cys-His dyads (e.g. hepatitis A virus protease), whilst in others the cysteine is already deprotonated before catalysis begins (e.g. papain).[29] This triad is also used by some amidases, such as N-glycanase to hydrolyse non-peptide C-N bonds.[30]
Ser-His-His
The triad of cytomegalovirus protease[lower-alpha 2] uses histidine as both the acid and base triad members. Removing the acid histidine results in only a 10-fold activity loss (compared to >10,000-fold when aspartate is removed from chymotrypsin). This triad has been interpreted as a possible way of generating a less active enzyme to control cleavage rate.[22]
Ser-Glu-Asp
An unusual triad is found in seldolisin proteases.[lower-alpha 6] The low pKa of the glutamate carboxylate group means that it only acts as a base in the triad at very low pH. The triad is hypothesised to be an adaptation to specific environments like acidic hot springs (e.g. kumamolysin) or cell lysosome (e.g. tripeptidyl peptidase).[22]
Thr-Nter, Ser-Nter and Cys-Nter
Threonine proteases, such as the proteasome protease subunit[lower-alpha 7] and ornithine acyltransferases[lower-alpha 8] use the secondary hydroxyl of threonine in a manner analogous to the use of the serine primary hydroxyl.[27][28] However, due to the steric interference of the extra methyl group of threonine, the base member of the triad is the N-terminal amide which polarises an ordered water which, in turn, deprotonates the catalytic hydroxyl to increase its reactivity.[1][22] Similarly, there exist equivalent 'serine only' and 'cysteine only' configurations such as penicillin acylase G[lower-alpha 9] and penicillin acylase V[lower-alpha 10] which are evolutionarily related to the proteasome proteases. Again, these use their N-terminal amide as a base.[22]
Ser-cisSer-Lys
This unusual triad occurs only in one superfamily of amidases. In this case, the lysine acts to polarise the middle serine. The middle serine then forms two strong hydrogen bonds to the nucleophilic serine to activate it (one with the side chain hydroxyl and the other with the backbone amide). The middle serine is held in an unusual cis orientation to facilitate precise contacts with the other two triad residues. The triad is further unusual in that the lysine and cis-serine both act as the base in activating the catalytic serine, but the same lysine also performs the role of the acid member as well as making key structural contacts.[31]
Divergent evolution
The sophistication of the active site network causes residues involved in catalysis (and residues in contact with these) to be highly evolutionarily conserved.[32] However, there are examples of divergent evolution in catalytic triads, both in the reaction catalysed, and the residues used in catalysis. The triad remains the core of the active site, but it is evolutionarily adapted to serve different functions.[33][34]
Reaction changes
Catalytic triads perform covalent catalysis via an acyl-enzyme intermediate. If this intermediate is resolved by water, the result is hydrolysis of the substrate. However, if the intermediate is resolved by attack by a second substrate, then the enzyme acts as a transferase. For example, attack by an acyl group results in an acyltransferase reaction. Several families of transferase enzymes have evolved from hydrolases by adaptation to exclude water and favour attack of a second substrate.[35]
Additionally, an alternative transferase mechanism has been evolved by amidophosphoribosyltransferases, which has two active sites.[lower-alpha 11] In the first active site, a cysteine triad hydrolyses a glutamine substrate to release free ammonia. The ammonia then diffuses though an internal tunnel in the enzyme to the second active site, where it is transferred to a second substrate.[36][37]
Nucleophile changes

Divergent evolution of active site residues is slow, due to strong chemical constraints. Nevertheless, some protease superfamilies have evolved from one nucleophile to another. This can be inferred when a superfamily (with the same fold) contains families that use different nucleophiles. Such nucleophile switches have occurred several times during evolutionary history, however the mechanisms by which this happen are still unclear.[17]
Within protease superfamilies that contain a mixture of nucleophiles (e.g. the PA clan), families are designated by their catalytic nucleophile (C=cysteine proteases, S=serine proteases).
| Superfamily | Families | Examples |
|---|---|---|
| PA clan | C3, C4, C24, C30, C37, C62, C74, C99 | TEV protease (Tobacco etch virus) |
| S1, S3, S6, S7, S29, S30, S31, S32, S39, S46, S55, S64, S65, S75 | Chymotrypsin (mammals, e.g. Bos taurus) | |
| PB clan | C44, C45, C59, C69, C89, C95 | Amidophosphoribosyltransferase precursor (Homo sapiens) |
| S45, S63 | Penicillin G acylase precursor (Escherichia coli) | |
| T1, T2, T3, T6 | Archaean proteasome, beta component (Thermoplasma acidophilum) | |
| PC clan | C26, C56 | Gamma-glutamyl hydrolase (Rattus norvegicus) |
| S51 | Dipeptidase E (Escherichia coli) | |
| PD clan | C46 | Hedgehog protein (Drosophila melanogaster) |
| N9, N10, N11 | Intein-containing V-type proton ATPase catalytic subunit A (Saccharomyces cerevisiae) | |
| PE clan | P1 | DmpA aminopeptidase (Ochrobactrum anthropi) |
| T5 | Ornithine acetyltransferase precursor (Saccharomyces cerevisiae) |
Convergent evolution


The enzymology of proteases provides some of the clearest known examples of convergent evolution. The same geometric arrangement of triad residues occurs in over 20 separate enzyme superfamilies. Each of these superfamilies is the result of convergent evolution for the same triad arrangement within a different structural fold. This is because there are limited productive ways to arrange three triad residues, the enzyme backbone and the substrate. These examples reflect the intrinsic chemical and physical constraints on enzymes, leading evolution to repeatedly and independently converge on equivalent solutions.[1][2]
Cysteine and serine hydrolases
The same triad geometries been converged upon by serine proteases such as the chymotrypsin[lower-alpha 5] and subtilisin superfamilies. Similar convergent evolution has occurred with cysteine proteases such as viral C3 protease and papain[lower-alpha 3] superfamilies. These triads have converged to almost the same arrangement due to the mechanistic similarities in cysteine and serine proteolysis mechanisms.[2]
Families of Cysteine proteases
| Superfamily | Families | Examples |
|---|---|---|
| CA | C1, C2, C6, C10, C12, C16, C19, C28, C31, C32, C33, C39, C47, C51, C54, C58, C64, C65, C66, C67, C70, C71, C76, C78, C83, C85, C86, C87, C93, C96, C98, C101 | Papain (Carica papaya) and calpain (Homo sapiens) |
| CD | C11, C13, C14, C25, C50, C80, C84 | Caspase-1 (Rattus norvegicus) and separase (Saccharomyces cerevisiae) |
| CE | C5, C48, C55, C57, C63, C79 | Adenain (human adenovirus type 2) |
| CF | C15 | Pyroglutamyl-peptidase I (Bacillus amyloliquefaciens) |
| CL | C60, C82 | Sortase A (Staphylococcus aureus) |
| CM | C18 | Hepatitis C virus peptidase 2 (hepatitis C virus) |
| CN | C9 | Sindbis virus-type nsP2 peptidase (sindbis virus) |
| CO | C40 | Dipeptidyl-peptidase VI (Lysinibacillus sphaericus) |
| CP | C97 | DeSI-1 peptidase (Mus musculus) |
| PA | C3, C4, C24, C30, C37, C62, C74, C99 | TEV protease (Tobacco etch virus) |
| PB | C44, C45, C59, C69, C89, C95 | Amidophosphoribosyltransferase precursor (Homo sapiens) |
| PC | C26, C56 | Gamma-glutamyl hydrolase (Rattus norvegicus) |
| PD | C46 | Hedgehog protein (Drosophila melanogaster) |
| PE | P1 | DmpA aminopeptidase (Ochrobactrum anthropi) |
| unassigned | C7, C8, C21, C23, C27, C36, C42, C53, C75 |
Families of Serine proteases
| Superfamily | Families | Examples |
|---|---|---|
| SB | S8, S53 | Subtilisin (Bacillus licheniformis) |
| SC | S9, S10, S15, S28, S33, S37 | Prolyl oligopeptidase (Sus scrofa) |
| SE | S11, S12, S13 | D-Ala-D-Ala peptidase C (Escherichia coli) |
| SF | S24, S26 | Signal peptidase I (Escherichia coli) |
| SH | S21, S73, S77, S78, S80 | Cytomegalovirus assemblin (human herpesvirus 5) |
| SJ | S16, S50, S69 | Lon-A peptidase (Escherichia coli) |
| SK | S14, S41, S49 | Clp protease (Escherichia coli) |
| SO | S74 | Phage GA-1 neck appendage CIMCD self-cleaving protein (Bacillus phage GA-1) |
| SP | S59 | Nucleoporin 145 (Homo sapiens) |
| SR | S60 | Lactoferrin (Homo sapiens) |
| SS | S66 | Murein tetrapeptidase LD-carboxypeptidase (Pseudomonas aeruginosa) |
| ST | S54 | Rhomboid-1 (Drosophila melanogaster) |
| PA | S1, S3, S6, S7, S29, S30, S31, S32, S39, S46, S55, S64, S65, S75 | Chymotrypsin A (Bos taurus) |
| PB | S45, S63 | Penicillin G acylase precursor (Escherichia coli) |
| PC | S51 | Dipeptidase E (Escherichia coli) |
| PE | P1 | DmpA aminopeptidase (Ochrobactrum anthropi) |
| unassigned | S48, S62, S68, S71, S72, S79, S81 |
Threonine proteases
Threonine proteases use the amino acid threonine as their catalytic nucleophile. Unlike cysteine and serine, threonine is a secondary hydroxyl (i.e. has a methyl group). This methyl group greatly restricts the possible orientations of triad and substrate as the methyl clashes with either the enzyme backbone or histidine base. Consequently, the catalytic residue of a threonine protease is located at it N-terminus.[2]
Two evolutionarily independent enzyme superfamilies with different protein folds are known to use the N-terminal residue as a nucleophile: Superfamily PB (proteasomes using the Ntn fold)[27] and Superfamily PE (acetyltransferases using the DOM fold)[28] This commonality of active site structure in completely different protein folds indicates that the active site evolved convergently in those superfamilies.[2][22]
Families of threonine proteases
| Superfamily | Families | Examples |
|---|---|---|
| PB clan | T1, T2, T3, T6 | Archaean proteasome, beta component (Thermoplasma acidophilum) |
| PE clan | T5 | Ornithine acetyltransferase (Saccharomyces cerevisiae) |
See also
References
Notes
- 1 2 3 4 TEV protease MEROPS: clan PA, family C4
- 1 2 Cytomegalovirus protease MEROPS: clan SH, family S21
- 1 2 3 4 Papain MEROPS: clan CA, family C1
- ↑ Hepatitus A virus protease MEROPS: clan PA, family C3
- 1 2 3 Chymotrypsin MEROPS: clan PA, family S1
- ↑ Seldolisin protease MEROPS: clan SB, family 53
- 1 2 Proteasome MEROPS: clan PB, family T1
- 1 2 Ornithine acyltransferases MEROPS: clan PE, family T5
- ↑ Penicillin acylase G MEROPS: clan PB, family S45
- ↑ Penicillin acylase V MEROPS: clan PB, family C59
- ↑ amidophosphoribosyltransferase MEROPS: clan PB, family C44
- ↑ Subtilisin MEROPS: clan SB, family S8
- ↑ Prolyl oligopeptidase MEROPS: clan SC, family S9
Citations
- 1 2 3 4 5 6 Dodson, G; Wlodawer, A (September 1998). "Catalytic triads and their relatives.". Trends in Biochemical Sciences. 23 (9): 347–52. PMID 9787641. doi:10.1016/S0968-0004(98)01254-7.
- 1 2 3 4 5 6 7 8 9 10 11 12 Buller, AR; Townsend, CA (Feb 19, 2013). "Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad". Proceedings of the National Academy of Sciences of the United States of America. 110 (8): E653–61. Bibcode:2013PNAS..110E.653B. PMC 3581919
 . PMID 23382230. doi:10.1073/pnas.1221050110.
. PMID 23382230. doi:10.1073/pnas.1221050110. - 1 2 3 4 5 6 Stryer L, Berg JM, Tymoczko JL (2002). "9 Catalytic Strategies". Biochemistry (5th ed.). San Francisco: W.H. Freeman. ISBN 0-7167-4955-6.
- 1 2 Perutz, Max (1992). Protein structure. New approaches to disease and therapy. New York: W.H. Freeman and Co.
- 1 2 Neurath, H (Oct 1994). "Proteolytic enzymes past and present: the second golden era. Recollections, special section in honor of Max Perutz". Protein Sci. 3 (10): 1734–9. PMC 2142620 . PMID 7849591. doi:10.1002/pro.5560031013.
- ↑ Ohman, KP; Hoffman, A; Keiser, HR (April 1990). "Endothelin-induced vasoconstriction and release of atrial natriuretic peptides in the rat.". Acta physiologica Scandinavica. 138 (4): 549–56. PMID 2141214. doi:10.1111/j.1748-1716.1990.tb08883.x.
- ↑ Dixon, Gordon H.; Kauffman, Dorothy L.; Neurath, Hans (5 March 1958). "Amino Acid Sequence in the Region of Diisopropyl Phosphoryl Binding in Dip-Trypsin". Journal of the American Chemical Society. 80 (5): 1260–1261. doi:10.1021/ja01538a059.
- ↑ Matthews BW, Sigler PB, Henderson R, Blow DM (1967). "Three-dimensional structure of tosyl-α-chymotrypsin". Nature. 214 (5089): 652–656. Bibcode:1967Natur.214..652M. doi:10.1038/214652a0.
- ↑ WALSH, KA; NEURATH, H (October 1964). "TRYPSINOGEN AND CHYMOTRYPSINOGEN AS HOMOLOGOUS PROTEINS". Proceedings of the National Academy of Sciences of the United States of America. 52 (4): 884–9. Bibcode:1964PNAS...52..884W. PMC 300366 . PMID 14224394. doi:10.1073/pnas.52.4.884.
- ↑ de Haën, C; Neurath, H; Teller, DC (Feb 25, 1975). "The phylogeny of trypsin-related serine proteases and their zymogens. New methods for the investigation of distant evolutionary relationships.". Journal of Molecular Biology. 92 (2): 225–59. PMID 1142424. doi:10.1016/0022-2836(75)90225-9.
- ↑ Lesk, AM; Fordham, WD (May 10, 1996). "Conservation and variability in the structures of serine proteinases of the chymotrypsin family.". Journal of Molecular Biology. 258 (3): 501–37. PMID 8642605. doi:10.1006/jmbi.1996.0264.
- ↑ Blow, DM; Birktoft, JJ; Hartley, BS (Jan 25, 1969). "Role of a buried acid group in the mechanism of action of chymotrypsin". Nature. 221 (5178): 337–40. Bibcode:1969Natur.221..337B. PMID 5764436. doi:10.1038/221337a0.
- ↑ Gorbalenya, AE; Blinov, VM; Donchenko, AP (Jan 6, 1986). "Poliovirus-encoded proteinase 3C: a possible evolutionary link between cellular serine and cysteine proteinase families.". FEBS Letters. 194 (2): 253–7. PMID 3000829. doi:10.1016/0014-5793(86)80095-3.
- ↑ Bazan, JF; Fletterick, RJ (November 1988). "Viral cysteine proteases are homologous to the trypsin-like family of serine proteases: structural and functional implications". Proceedings of the National Academy of Sciences of the United States of America. 85 (21): 7872–6. Bibcode:1988PNAS...85.7872B. PMC 282299 . PMID 3186696. doi:10.1073/pnas.85.21.7872.
- ↑ Phan, J; Zdanov, A; Evdokimov, AG; Tropea, JE; Peters HK, 3rd; Kapust, RB; Li, M; Wlodawer, A; Waugh, DS (Dec 27, 2002). "Structural basis for the substrate specificity of tobacco etch virus protease". The Journal of Biological Chemistry. 277 (52): 50564–72. PMID 12377789. doi:10.1074/jbc.M207224200.
- ↑ Rawlings, N.D.; Barrett, A.J. (1993). "Evolutionary families of peptidases". Biochem J. 290: 205–218.
- 1 2 Rawlings ND, Barrett AJ, Bateman A (January 2010). "MEROPS: the peptidase database". Nucleic Acids Res. 38 (Database issue): D227–33. PMC 2808883 . PMID 19892822. doi:10.1093/nar/gkp971.
- ↑ Frey, P. A.; Whitt, S. A.; Tobin, J. B. (1994-06-24). "A low-barrier hydrogen bond in the catalytic triad of serine proteases". Science. 264 (5167): 1927–1930. Bibcode:1994Sci...264.1927F. ISSN 0036-8075. PMID 7661899. doi:10.1126/science.7661899.
- ↑ Ash, Elissa L.; Sudmeier, James L.; Fabo, Edward C. De; Bachovchin, William W. (1997-11-07). "A Low-Barrier Hydrogen Bond in the Catalytic Triad of Serine Proteases? Theory Versus Experiment". Science. 278 (5340): 1128–1132. Bibcode:1997Sci...278.1128A. ISSN 0036-8075. PMID 9353195. doi:10.1126/science.278.5340.1128.
- ↑ Schutz, Claudia N.; Warshel, Arieh (2004-01-01). "The low barrier hydrogen bond (LBHB) proposal revisited: The case of the Asp ··· His pair in serine proteases". Proteins: Structure, Function, and Bioinformatics. 55 (3): 711–723. doi:10.1002/prot.20096.
- ↑ Warshel, Arieh; Papazyan, Arno (1996-11-26). "Energy considerations show that low-barrier hydrogen bonds do not offer a catalytic advantage over ordinary hydrogen bonds". Proceedings of the National Academy of Sciences. 93 (24): 13665–13670. Bibcode:1996PNAS...9313665W. ISSN 0027-8424. PMC 19385 . PMID 8942991. doi:10.1073/pnas.93.24.13665.
- 1 2 3 4 5 6 Ekici, OD; Paetzel, M; Dalbey, RE (December 2008). "Unconventional serine proteases: variations on the catalytic Ser/His/Asp triad configuration.". Protein science : a publication of the Protein Society. 17 (12): 2023–37. PMC 2590910 . PMID 18824507. doi:10.1110/ps.035436.108.
- 1 2 McGrath, ME; Wilke, ME; Higaki, JN; Craik, CS; Fletterick, RJ (Nov 28, 1989). "Crystal structures of two engineered thiol trypsins.". Biochemistry. 28 (24): 9264–70. PMID 2611228. doi:10.1021/bi00450a005.
- 1 2 Polgár, L; Asbóth, B (Aug 7, 1986). "The basic difference in catalyses by serine and cysteine proteinases resides in charge stabilization in the transition state.". Journal of Theoretical Biology. 121 (3): 323–6. PMID 3540454. doi:10.1016/s0022-5193(86)80111-4.
- ↑ Damblon, C; Raquet, X; Lian, LY; Lamotte-Brasseur, J; Fonze, E; Charlier, P; Roberts, GC; Frère, JM (Mar 5, 1996). "The catalytic mechanism of beta-lactamases: NMR titration of an active-site lysine residue of the TEM-1 enzyme". Proceedings of the National Academy of Sciences of the United States of America. 93 (5): 1747–52. Bibcode:1996PNAS...93.1747D. PMC 39852 . PMID 8700829. doi:10.1073/pnas.93.5.1747.
- ↑ Jelsch, C; Lenfant, F; Masson, JM; Samama, JP (Mar 9, 1992). "Beta-lactamase TEM1 of E. coli. Crystal structure determination at 2.5 A resolution.". FEBS Letters. 299 (2): 135–42. PMID 1544485. doi:10.1016/0014-5793(92)80232-6.
- 1 2 3 Brannigan, JA; Dodson, G; Duggleby, HJ; Moody, PC; Smith, JL; Tomchick, DR; Murzin, AG (Nov 23, 1995). "A protein catalytic framework with an N-terminal nucleophile is capable of self-activation". Nature. 378 (6555): 416–9. Bibcode:1995Natur.378..416B. PMID 7477383. doi:10.1038/378416a0.
- 1 2 3 Cheng, H; Grishin, NV (July 2005). "DOM-fold: a structure with crossing loops found in DmpA, ornithine acetyltransferase, and molybdenum cofactor-binding domain.". Protein science : a publication of the Protein Society. 14 (7): 1902–10. PMC 2253344 . PMID 15937278. doi:10.1110/ps.051364905.
- ↑ Beveridge, AJ (July 1996). "A theoretical study of the active sites of papain and S195C rat trypsin: implications for the low reactivity of mutant serine proteinases". Protein science : a publication of the Protein Society. 5 (7): 1355–65. PMC 2143470 . PMID 8819168. doi:10.1002/pro.5560050714.
- ↑ Allen, Mark D.; Buchberger, Alexander; Bycroft, Mark (2006). "The PUB Domain Functions as a p97 Binding Module in Human Peptide N-Glycanase". Journal of Biological Chemistry. 281 (35): 25502–25508. ISSN 0021-9258. PMID 16807242. doi:10.1074/jbc.M601173200.
- ↑ Shin, S; Yun, YS; Koo, HM; Kim, YS; Choi, KY; Oh, BH (Jul 4, 2003). "Characterization of a novel Ser-cisSer-Lys catalytic triad in comparison with the classical Ser-His-Asp triad.". The Journal of Biological Chemistry. 278 (27): 24937–43. PMID 12711609. doi:10.1074/jbc.M302156200.
- ↑ Halabi, N; Rivoire, O; Leibler, S; Ranganathan, R (Aug 21, 2009). "Protein sectors: evolutionary units of three-dimensional structure.". Cell. 138 (4): 774–86. PMC 3210731 . PMID 19703402. doi:10.1016/j.cell.2009.07.038.
- ↑ Murzin, Alexey G (June 1998). "How far divergent evolution goes in proteins". Current Opinion in Structural Biology. 8 (3): 380–387. doi:10.1016/S0959-440X(98)80073-0.
- ↑ Gerlt, John A.; Babbitt, Patricia C. (2001). "Divergent Evolution of Enzymatic Function: Mechanistically Diverse Superfamilies and Functionally Distinct Suprafamilies". Annual Review of Biochemistry. 70 (1): 209–246. PMID 11395407. doi:10.1146/annurev.biochem.70.1.209.
- ↑ Stehle, Felix; Brandt, Wolfgang; Stubbs, Milton T.; Milkowski, Carsten; Strack, Dieter (October 2009). "Sinapoyltransferases in the light of molecular evolution". Phytochemistry. Evolution of Metabolic Diversity. 70 (15–16): 1652–1662. PMID 19695650. doi:10.1016/j.phytochem.2009.07.023.
- ↑ Smith JL (Dec 1998). "Glutamine PRPP amidotransferase: snapshots of an enzyme in action". Current Opinion in Structural Biology. 8 (6): 686–94. PMID 9914248. doi:10.1016/s0959-440x(98)80087-0.
- ↑ Smith JL, Zaluzec EJ, Wery JP, Niu L, Switzer RL, Zalkin H, Satow Y (Jun 1994). "Structure of the allosteric regulatory enzyme of purine biosynthesis". Science. 264 (5164): 1427–1433. Bibcode:1994Sci...264.1427S. PMID 8197456. doi:10.1126/science.8197456.