Carbonyl alpha-substitution reactions
Alpha-substitution reactions occur at the position next to the carbonyl group, the α-position, and involve the substitution of an α hydrogen atom by an electrophile, E, through either an enol or enolate ion intermediate.[1]

Reaction mechanism
Because their double bonds are electron rich, enols behave as nucleophiles and react with electrophiles in much the same way that alkenes do. But because of resonance electron donation of a lonepair of electron s on the neighboring oxygen, enols are more electron- rich and correspondingly more reactive than alkenes. Notice in the following electrostatic potential map of ethenol (H2C=CHOH) how there is a substantial amount of electron density on the α carbon.

When an alkene reacts with an electrophile, such as HCl, initial addition of H+ gives an intermediate cation and subsequent reaction with Cl− yields an addition product. When an enol reacts with an electrophile, however, only the initial addition step is the same. Instead of reacting with CI− to give an addition product, the intermediate cation loses the OH− proton to give an α-substituted carbonyl compound.[1]:845
Alpha-halogenation of aldehydes and ketones
A particularly common α-substitution reaction in the laboratory is the halogenation of aldehydes and ketones at their α positions by reaction Cl2, Br2 or I2 in acidic solution. Bromine in acetic acid solvent is often used.
Remarkably, ketone halogenation also occurs in biological systems, particularly in marine alga, where dibromoacetaldehyde, bromoacetone, 1, l,l -tribromoacetone, and other related compounds have been found.
The halogenation is a typical α-substitution reaction that proceeds by acid catalyzed formation of an enol intermediate.[1]:846
Acidity of alpha-hydrogen atoms: enolate ion formation
A hydrogen on the α position of a carbonyl compound is weakly acidic and can be removed by a strong base to yield an enolate ion. In comparing acetone (pKa= 19.3) with ethane (pKa= 60), for instance, the presence of a neighboring carbonyl group increases the acidity of the ketone over the alkane by a factor of 1040.
Abstraction of a proton from a carbonyl compound occurs when the a C-H bond is oriented roughly parallel to the p orbitals of the carbonyl group. The α carbon atom of the enolate ion is sp2-hybridized and has a p orbital that overlaps the neighboring carbonyl p orbitals. Thus, the negative charge is shared by the electronegative oxygen atom, and the enolate ion is stabilized by resonance.
Carbonyl compounds are more acidic than alkanes for the same reason that carboxylic acids are more acidic than alcohols. In both cases, the anions are stabilized by resonance. Enolate ions differ from carboxylate ions, however, in that their two resonance forms are not equivalent- the form with the negative charge on oxygen is lower in energy than the form with the charge on carbon. Nevertheless, the principle behind resonance stabilization is the same in both cases.
Because carbonyl compounds are only weakly acidic, a strong base is needed for enolate ion formation . If an alkoxide such as sodium ethoxide is used as base, deprotonation takes place only to the extent of about 0.1% because acetone is a weaker acid than ethanol (pKa= 16). If, however, a more powerful base such as sodium hydride (NaH) or lithium diisopropylamide (LDA) is used, a carbonyl compound can be completely converted into its enolate ion. Lithium diisopropylamide (LDA), which is easily prepared by reaction of the strong base butyllithium with diisopropylamine, is widely used in the laboratory as a base for preparing enolate ions from carbonyl compounds.
Many types of carbonyl compounds, including aldehydes, ketones, esters, thioesters, acids, and amides, can be converted into enolate ions by reaction with LDA. Note that nitriles, too, are acidic and can be converted into enolate-like anions (referred to as nitrile anions). When a hydrogen atom is flanked by two carbonyl groups, its acidity is enhanced even more. This enhanced acidity of β-dicarbonyl compounds is due to the stabilization of the resultant enolate ions by delocalization of the negative charge over both carbonyl groups.[1]:850
Reactivity of enolate ions
Enolate ions are more useful than enols for two reasons. First, pure enols can't normally be isolated but are instead generated only as short lived intermediates in low concentration. By contrast, stable solutions of pure enolate ions are easily prepared from most carbonyl compounds by reaction with a strong base. Second, enolate ions are more reactive than enols and undergo many reactions that enols don't. Whereas enols are neutral, enolate ions are negatively charged, making them much better nucleophiles. As a result, enolate ions are more common than enols in both laboratory and biological chemistry.
Because they are resonance hybrids of two nonequivalent forms, enolate ions can be looked at either as vinylic alkoxides (C=C- O−) or as α-ketocarbanions (−C-C= O). Thus, enolate ions can react with electrophiles either on oxygen or on carbon. Reaction on oxygen yields an enol derivative, while reaction on carbon yields an α-substituted carbonyl compound. Both kinds of reactivity are known, but reaction on carbon is more common.[1]:853
Alkylation of enolate ions
Perhaps the single most important reaction of enolate ions is their alkylation by treatment with an alkyl halide or tosylate, thereby forming a new C-C bond and joining two smaller pieces into one larger molecule. Alkylation occurs when the nucleophilic enolate ion reacts with the electrophilic alkyl halide in an SN2 reaction and displaces the leaving group by backside attack.
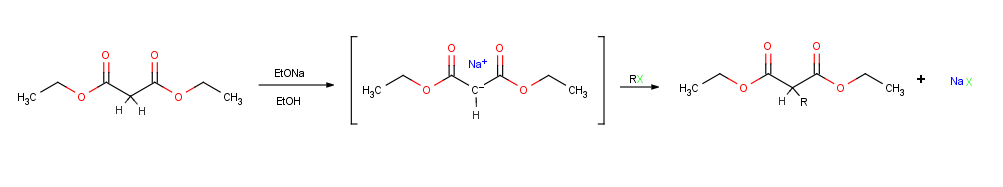
Alkylation reactions are subject to the same constraints that affect all SN2 reactions. Thus, the leaving group X in the alkylating agent R-X can be chloride, bromide, iodide, or tosylate . Tile alkyl group R should be primary or methyl, and preferably should be allylic or benzylic. Secondary halides react poorly, and tertiary halides don't react at all because a competing E2 elimination of HX occurs instead. Vinylic and aryl halides are also unreactive because backside approach is sterically prevented.[1]:855