CLIP
CLIP (cross-linking immunoprecipitation) is a method used in molecular biology that combines UV cross-linking with immunoprecipitation in order to analyse protein interactions with RNA or to precisely locate RNA modifications (e.g. m6A).[1][2][3][4] CLIP-based techniques can be used to map RNA binding protein binding sites or RNA modification sites [4][5] of interest on a genome-wide scale, thereby increasing the understanding of post-transcriptional regulatory networks.
Workflow
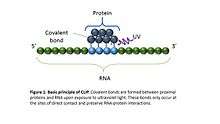
CLIP begins with the in-vivo cross-linking of RNA-protein complexes using ultraviolet light (UV). Upon UV exposure, covalent bonds are formed between proteins and nucleic acids that are in close proximity.[6] The cross-linked cells are then lysed, and the protein of interest is isolated via immunoprecipitation. In order to allow for sequence specific priming of reverse transcription, RNA adapters are ligated to the 3' ends, while radiolabeled phosphates are transferred to the 5' ends of the RNA fragments. The RNA-protein complexes are then separated from free RNA using gel electrophoresis and membrane transfer. Proteinase K digestion is then performed in order to remove protein from the RNA-protein complexes. This step leaves a peptide at the cross-link site, allowing for the identification of the cross-linked nucleotide.[7] After ligating RNA linkers to the RNA 5' ends, cDNA is synthesized via RT-PCR. High-throughput sequencing is then used to generate reads containing distinct barcodes that identify the last cDNA nucleotide. Interaction sites can be identified by mapping the reads back to the transcriptome.
History and applications
CLIP was originally undertaken to study interactions between the neuron-specific RNA-binding protein and splicing factor NOVA1 and NOVA2 in the mouse brain, identifying RNA binding sites that had Nova binding sites and were validated as Nova targets in knock-out mouse brains.[8] In 2008 CLIP was combined with high-throughput sequencing (termed "HITS-CLIP") to generate genome-wide protein-RNA interaction maps for Nova;[9] since then a number of other splicing factor maps have been generated, including those for PTB,[10] RbFox2 (where it was renamed "CLIP-seq"),[11] SFRS1,[12] Argonaute,[13] hnRNP C,[14] the Fragile-X mental retardation protein FMRP,[15] Ptbp2 (in the mouse brain),[16] Mbnl2,[17] the nElavl proteins (the neuron-specific Hu proteins),[18] and even N6-Methyladenosine(m6A) RNA modification antibody.[4] A review of the range of proteins studied by HITS-CLIP has been published.[19]
HITS-CLIP (CLIP-seq) analysis of the RNA-binding protein Argonaute has been performed for the identification of microRNA targets[20] by decoding microRNA-mRNA and protein-RNA interaction maps in the mouse brain,[13][21] and subsequently in Caenorhabditis elegans,[22] embryonic stem cells,[23] and tissue culture cells.[24] As a novel modification of HITS-CLIP, m6A-CLIP was developed to precisely map m6A locations in mRNA by UV-crosslinking m6A antibody to the target RNA.[4] Recently, improved bioinformatics methods applied to Argonaute HITS-CLIP, have identified binding sites with single nucleotide resolution.[3]
miRNA target detection
The main steps (using Degradome sequencing concurrently) are:
- mapping CLIP-seq reads
- mapping Degradome-Seq reads
- grouping overlapping reads into clusters
- querying miRNA targets from different public databases
- identifying miRNA–target interactions with an alignment score from CleaveLand not exceeding the cutoff threshold of 7.0
- the ClipSearch program was developed to search for 6–8-mers (8-mer, 7-mer-m8 and 7-mer-A1) (2,5) in CLIP-Seq data
- The DegradomeSearch program was developed to search Degradome-Seq clusters for nearly perfect complements of miRNA sequences
Methods
HITS-CLIP or CLIP-Seq
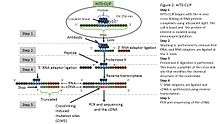
HITS-CLIP,[19] also known as CLIP-Seq, combines UV cross-linking and immunoprecipitation with high-throughput sequencing to identify binding sites of RNA-binding proteins. CLIP-seq depends on cross-linking induced mutation sites (CIMS) to localized protein-RNA binding sites.[3] Because CIMS are reproducible, high sequencing depths allow CIMS to be differentiated from technical errors.
PAR-CLIP

PAR-CLIP (photoactivatable ribonucleoside–enhanced cross-linking and immunoprecipitation) is a biochemical method used for identifying the binding sites of cellular RNA-binding proteins (RBPs) and microRNA-containing ribonucleoprotein complexes (miRNPs).[24] The method relies on the incorporation of photoreactive ribonucleoside analogs, such as 4-thiouridine (4-SU) and 6-thioguanosine (6-SG) into nascent RNA transcripts by living cells. Irradiation of the cells by UV light of 365 nm induces efficient cross-linking of photoreactive nucleoside-labeled cellular RNAs to interacting RBPs. Immunoprecipitation of the RBP of interest is followed by the isolation of the cross-linked and co-immunoprecipitated RNA. The isolated RNA is converted into a cDNA library and deep sequenced using high-throughput sequencing technology.[24][26] Cross-linking the 4-SU and 6-SG analogs results in thymidine to cytidine, and guanosine to adenosine transitions respectively. As a result, PAR-CLIP can identify binding site locations with high accuracy.[3]
However, PAR-CLIP is limited to cultured cells,[3][25] and nucleoside cytotoxicity is a concern;[2][24] it has been reported that 4-SU inhibits ribosomal RNA synthesis, induces a nucleolar stress response, and reduces cell proliferation.[27] It should be noted that 4-SU substitution occurs in approximately 1 out of every 40 uridine nucleosides, and that T to C transitions frequently occur at the cross-link site.[24]
Recently, PAR-CLIP has been employed to determine the transcriptome-wide binding sites of several known RBPs and microRNA-containing ribonucleoprotein complexes at high resolution. This includes the miRNA targeting AGO and TNRC6 proteins.[21][24]
iCLIP
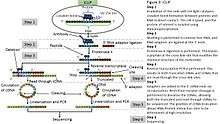
iCLIP (individual nucleotide–resolution cross-linking and immunoprecipitation) is a technique used for identifying protein-RNA interactions. The method uses UV light to covalently bind proteins and RNA molecules. As with all CLIP methods, iCLIP allows for the stringent purification of linked protein-RNA complexes, using immunoprecipitation followed by SDS-PAGE and membrane transfer. The radiolabelled protein-RNA complexes are then excised from the membrane, and treated with proteinase to release the RNA. This leaves one or two amino acids at the RNA cross-link site. The RNA is then reverse transcribed using barcoded primers. Because reverse transcription stops prematurely at the cross-link site, iCLIP allows RNA-protein interaction sites to be identified at high resolution. As a novel modification of iCLIP, m6A-CLIP further took advantage of the m6A-induced truncation sites (MITS) to precisely map m6A sites in mRNAs.[4]
Other CLIP methods
sCLIP (simple CLIP) is a technique that requires lower amounts of input RNA and omits radio-labeling of the immunoprecipitated RNA. The method is based on linear amplification of the immunoprecipitated RNA and thereby improves the complexity of the sequencing-library despite significantly reducing the amount of input material and omitting several purification steps. Additionally, it permits a radiolabel-free visualization of immunoprecipitated RNA by using a highly sensitive biotin-based labeling technique. Along with a bioinformatical platform this method is designed to provide deep insights into RNA–protein interactomes in biomedical science, where the amount of starting material is often limited (i.e. in case of precious clinical samples).[28]
Advantages and limitations
Advantages
Early methods for identifying RNA-protein interactions relied on either the affinity purification of RNA-binding proteins or the immunoprecipitation of RNA-protein complexes. These methods lacked a cross-linking step and obtained low signal to noise ratios.[8] Because RNA-binding proteins are frequently components of multi-protein complexes, RNAs bound to non-target proteins may be co-precipitated. The data obtained using early immunoprecipitation methods have been demonstrated to be dependent on the reaction conditions of the experiment. For example, the subset of RNA-protein interactions preserved depends highly on the protein concentrations and ionic conditions. Furthermore, the reassociation of RNA-binding proteins following cell lysis may lead to the detection of artificial interactions.[29]
Formaldehyde cross-linking methods have been used to preserve RNA-protein interactions, but also generate protein-protein cross-links. UV cross-linking methods provide a significant advantage over formaldehyde cross-linking, as they avoid protein-protein cross-links entirely. Proteinase K digestion also confers an advantage to CLIP methods due to the peptide left at the cross-link site. Reverse transcription of the fragments through the cross-link site introduces mutations that are specific to each separate CLIP method, and may be used to determine the binding site with high accuracy.[3]
Limitations

All CLIP library generation protocols require moderate quantities of cells or tissue (50–100 mg), require numerous enzymatic steps, and, for HITS-CLIP, extensive informatic analysis (as recently reviewed).[30] Certain steps are difficult to optimize and frequently have low efficiencies. For example, overdigestion with RNase can decrease the number of identified binding sites.[25] Cross-linking also presents a concern. The optimal cross-linking protocol varies between proteins,[8] and the efficiency is typically between 1-5%. Cross-linking bias has been reported in the literature,[31] but the impact of biases present in CLIP methods remains debatable. Computationally predicted miRNA targets derived from TargetScan[32] are comparable to CLIP in identifying miRNA targets, raising questions as to its utility relative to existing predictions.[32] Because CLIP methods rely on immunoprecipitation, antibody-epitope interactions are a potential obstacle. For instance, cross-linking at the epitope could impede antibody binding. Finally, significant differences have been observed between cross-linking sites in vivo in living cells and in vitro.[33] Therefore, CLIP results may not necessarily reflect RNA-protein binding site interactions within the cell.
Similar methods
- RIP-Chip, same goal and first steps, but doesn't use cross-linking and uses microarray instead of sequencing
- ChIP-Seq, for finding interactions with DNA rather than RNA
- SELEX, a method for finding a consensus binding sequence
Further reading
- starBase database: a database for exploring miRNA-lncRNA, miRNA-mRNA, miRNA-sncRNA, miRNA-circRNA, protein-lncRNA, protein-RNA interactions and ceRNA networks from PAR-CLIP'(CLIP-Seq, HITS-CLIP'iCLIP, CLASH) data, and TargetScan,[32] PicTar, RNA22, miRanda and PITA microRNA target sites.
- BIMSB doRiNA database: a database for exploring protein-RNA and microRNA-target interactions from CLIP-Seq, HITS-CLIP, PAR-CLIP, iCLIP data and PICTAR microRNA target site predictions.
- miRTarCLIP: A computational approach for identifying microRNA-target interactions using high-throughput CLIP and PAR-CLIP sequencing.
- clipz: a pipeline to analyze short RNA reads from HITS-CLIP experiments.
- dCLIP: dCLIP is a Perl program for discovering differential binding regions in two comparative CLIP-Seq (HITS-CLIP, PAR-CLIP or iCLIP) experiments.
References
- ↑ Ule et al. 2003.
- 1 2 3 4 5 6 Sugimoto et al. 2012.
- 1 2 3 4 5 6 7 Zhang & Darnell 2011.
- 1 2 3 4 5 Ke et al. 2015.
- ↑ Ke et al. 2017.
- ↑ Darnell 2012.
- ↑ König, McGlincy & Ule 2012.
- 1 2 3 Ule et al. 2005.
- ↑ Licatalosi et al. 2008.
- ↑ Xue et al. 2009.
- ↑ Yeo et al. 2009.
- ↑ Sanford et al. 2009.
- 1 2 Chi et al. 2009.
- ↑ König et al. 2010.
- ↑ Darnell et al. 2011.
- ↑ Licatalosi et al. 2012.
- ↑ Charizanis et al. 2012.
- ↑ Ince-Dunn et al. 2012.
- 1 2 Darnell 2010.
- ↑ Thomson, Bracken & Goodall 2011.
- 1 2 Yang et al. 2011.
- ↑ Zisoulis et al. 2010.
- ↑ Leung et al. 2011.
- 1 2 3 4 5 6 Hafner et al. 2010.
- 1 2 3 4 5 6 König et al. 2012.
- ↑ Hafner et al. 2010b.
- ↑ Burger et al. 2013.
- ↑ Kargapolova et al. 2017.
- ↑ Mili & Steitz 2004.
- ↑ Moore et al. 2014.
- ↑ Fecko et al. 2007.
- 1 2 3 Agarwal et al. 2015.
- ↑ Bohnsack et al. 2009.
Sources
- Agarwal, V; Bell, GW; Nam, J-W; Bartel, DP (2015). "Predicting effective microRNA target sites in mammalian mRNAs". eLife. 4: e05005. PMC 4532895
 . PMID 26267216. doi:10.7554/eLife.05005.
. PMID 26267216. doi:10.7554/eLife.05005. - Bohnsack, MT; Martin, R; Granneman, S; Ruprecht, M; Schleiff, E; Tollervey, D (2009). "Prp43 bound at different sites on the pre-rRNA performs distinct functions in ribosome synthesis". Molecular Cell. 36 (4): 583–592. PMC 2806949 . PMID 19941819. doi:10.1016/j.molcel.2009.09.039.
- Burger, K; Mühl, B; Kellner, M; Rohrmoser, M; Gruber-Eber, A; Windhager, L; Friedel, CC; Dölken, L; Eick, D (2013). "4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response". RNA Biol. 10 (10): 1623–1630. PMID 24025460. doi:10.4161/rna.26214.
- Charizanis, K; Lee, KY; Batra, R; Goodwin, M; Zhang, C; Yuan, Y; Shiue, L; Cline, M; Scotti, MM; Xia, G; Kumar, A; Ashizawa, T; Clark, HB; Kimura, T; Takahashi, MP; Fujimura, H; Jinnai, K; Yoshikawa, H; Gomes–Pereira, M; Gourdon, G; Sakai, N; Nishino, S; Foster, TC; Ares, M; Darnell, RB; Swanson, MS (2012). "Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy". Neuron. 75 (3): 437–450. PMC 3418517 . PMID 22884328. doi:10.1016/j.neuron.2012.05.029.
- Chi, SW; Zang, JB; Mele, A; Darnell, RB (2009). "Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps". Nature. 460 (7254): 479–486. PMC 2733940 . PMID 19536157. doi:10.1038/nature08170.
- Darnell, JC; Van Driesche, SJ; Zhang, C; Hung, KY; Mele, A; Fraser, CE; Stone, EF; Chen, C; Fak, JJ; Chi, SW; Licatalosi, DD; Richter, JD; Darnell, RB (2011). "FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism". Cell. 146 (2): 247–261. PMC 3232425 . PMID 21784246. doi:10.1016/j.cell.2011.06.013.
- Darnell, RB (2010). "HITS-CLIP: panoramic views of protein-RNA regulation in living cells". Wiley Interdiscip Rev RNA. 1 (2): 266–286. PMC 3222227 . PMID 21935890. doi:10.1002/wrna.31.
- Darnell, RB (2012). "CLIP (Cross-Linking and Immunoprecipitation) Identification of RNAs Bound by a Specific Protein". Cold Spring Harbor Protocols. 2012 (11): pdb.prot072132. doi:10.1101/pdb.prot072132.
- Fecko, CJ; Munson, KM; Saunders, A; Sun, G; Begley, TP; Lis, JT; Webb, WW (2007). "Comparison of femtosecond laser and continuous wave UV sources for protein-nucleic acid crosslinking". Photochemistry and Photobiology. 83 (6): 1394–1404. PMID 18028214. doi:10.1111/j.1751-1097.2007.00179.x.
- Hafner, M; Landthaler, M; Burger, L; Khorshid, M; Hausser, J; Berninger, P; Rothballer, A; Ascano, M; Jungkamp, AC; Munschauer, M; Ulrich, A; Wardle, GS; Dewell, S; Zavolan, M; Tuschl, T (2010). "Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP". Cell. 141 (1): 129–141. PMC 2861495 . PMID 20371350. doi:10.1016/j.cell.2010.03.009.
- Hafner, M; Landthaler, M; Burger, L; Khorshid, M; Hausser, J; Berninger, P; Rothballer, A; Ascano, M; Jungkamp, AC; Munschauer, M; Ulrich, A; Wardle, GS; Dewell, S; Zavolan, M; Tuschl, T (2010b). "PAR-CliP - A Method to Identify Transcriptome-wide the Binding Sites of RNA Binding Proteins". Journal of Visualized Experiments (41): e2034. PMC 3156069 . PMID 20644507. doi:10.3791/2034.
- Ince-Dunn, G; Okano, HJ; Jensen, KB; Park, WY; Zhong, R; Ule, J; Mele, A; Fak, JJ; Yang, CW; Zhang, C; Yoo, J; Herre, M; Okano, H; Noebels, JL; Darnell, RB (2012). "Neuronal Elav-like (Hu) proteins regulate RNA splicing and abundance to control glutamate levels and neuronal excitability". Neuron. 75 (6): 1067–1080. PMC 3517991 . PMID 22998874. doi:10.1016/j.neuron.2012.07.009.
- Ke, S; Alemu, EA; Mertens, C; Gantman, EC; Fak, JJ; Mele, A; Haripal, B; Zucker-Scharff, I; Moore, MJ; Park, CY; Vågbø, CB; Kusnierczyk, A; Klungland, A; Darnell, JE; Darnell, RB (2015). "A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation". Genes & Development. 29 (19): 2037–53. PMC 4604345 . PMID 26404942. doi:10.1101/gad.269415.115.
- Ke, S; Pandya-Jones, A; Saito, Y; Fak, JJ; Vågbø, CB; Geula, S; Hanna, JH; Black, DL; Darnell, JE; Darnell, RB (2017). "m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover". Genes & Development. 31 (10): 990–1006. PMID 28637692. doi:10.1101/gad.301036.117.
- König, J; Zarnack, K; Rot, G; Curk, T; Kayikci, M; Zupan, B; Turner, DJ; Luscombe, NM; Ule, J (2010). "iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution". Nat Struct Mol Biol. 17 (7): 909–915. PMC 3000544 . PMID 20601959. doi:10.1038/nsmb.1838.
- König, J; McGlincy, NJ; Ule, J (2012). "Analysis of Protein-RNA Interactions with Single-Nucleotide Resolution Using iCLIP and Next-Generation Sequencing". Tag-Based Next Generation Sequencing. p. 153. ISBN 978-3527644582. doi:10.1002/9783527644582.ch10.
- König, J; Zarnack, K; Luscombe, NM; Ule, J (2012). "Protein-RNA interactions: new genomic technologies and perspectives". Nature Reviews Genetics. 13 (2): 77–83. PMID 22251872. doi:10.1038/nrg3141.
- Leung, AK; Young, AG; Bhutkar, A; Zheng, GX; Bosson, AD; Nielsen, CB; Sharp, PA (2011). "Genome-wide identification of Ago2 binding sites from mouse embryonic stem cells with and without mature microRNAs". Nat Struct Mol Biol. 19 (9): 1084. doi:10.1038/nsmb0911-1084a.
- Licatalosi, DD; Mele, A; Fak, JJ; Ule, J; Kayikci, M; Chi, SW; Clark, TA; Schweitzer, AC; Blume, JE; Wang, X; Darnell, JC; Darnell, RB (2008). "HITS-CLIP yields genome-wide insights into brain alternative RNA processing". Nature. 456 (7221): 464–469. PMC 2597294 . PMID 18978773. doi:10.1038/nature07488.
- Licatalosi, DD; Yano, M; Fak, JJ; Mele, A; Grabinski, SE; Zhang, C; Darnell, RB (2012). "Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain". Genes Dev. 26 (14): 1626–1642. PMC 3404389 . PMID 22802532. doi:10.1101/gad.191338.112.
- Mili, S; Steitz, JA (2004). "Evidence for reassociation of RNA-binding proteins after cell lysis: Implications for the interpretation of immunoprecipitation analyses". RNA. 10 (11): 1692–4. PMC 1370654 . PMID 15388877. doi:10.1261/rna.7151404.
- Moore, JJ; Zhang, C; Gantman, EC; Mele, A; Darnell, JC; Darnell, RB (2014). "Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis". Nat Protocols. 9 (2): 263–293. PMC 4156013 . PMID 24407355. doi:10.1038/nprot.2014.012.
- Sanford, JR; Wang, X; Mort, M; Fanduyn, N; Cooper, DN; Mooney, SD; Edenberg, HJ; Liu, Y (2009). "Splicing factor SFRS1 recognizes a functionally diverse landscape of RNA transcripts". Genome Research. 19 (3): 381–394. PMC 2661799 . PMID 19116412. doi:10.1101/gr.082503.108.
- Sugimoto, Y; König, J; Hussain, S; Zupan, B; Curk, T; Frye, M; Ule, J (Aug 3, 2012). "Analysis of CLIP and iCLIP methods for nucleotide-resolution studies of protein-RNA interactions". Genome Biology. 13 (8): R67. PMC 4053741 . PMID 22863408. doi:10.1186/gb-2012-13-8-r67.
- Thomson, DW; Bracken, CP; Goodall, GJ (2011). "Experimental strategies for microRNA target identification". Nucleic Acids Research. 39 (16): 6845–6853. PMC 3167600 . PMID 21652644. doi:10.1093/nar/gkr330.
- Ule, J; Jensen, K; Ruggiu, M; Mele, A; Ule, A; Darnell, RB (Nov 14, 2003). "CLIP identified Nova-regulated RNA networks in the brain". Science. 302 (5648): 1212–1215. PMID 14615540. doi:10.1126/science.1090095.
- Ule, J; Jensen, K; Mele, A; Darnell, RB (2005). "CLIP: a method for identifying protein-RNA interactions sites in living cells". Methods. 37 (4): 376–386. PMID 16314267. doi:10.1016/j.ymeth.2005.07.018.
- Xue, Y; Zhou, Y; Wu, T; Zhu, T; Ju, X; Kwon, YS; Zhang, C; Yeo, G; Black, DL; Sun, H; Fu, XD; Zhang, Y (2009). "Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping". Molecular Cell. 36 (6): 996–1006. PMC 2807993 . PMID 20064465. doi:10.1016/j.molcel.2009.12.003.
- Yang, JH; Li, JH; Shao, P; Zhou, H; Chen, YQ; Qu, LH (2011). "starBase: a database for exploring microRNA–mRNA interaction maps from Argonaute CLIP-Seq and Degradome-Seq data". Nucl. Acids Res. 39 (Database issue): D202–D209. PMC 3013664 . PMID 21037263. doi:10.1093/nar/gkq1056.
- Yeo, GW; Coufal, NG; Liang, TY; Peng, GE; Fu, XD; Gage, FH (2009). "An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells". Nat Struct Mol Biol. 16 (2): 130–137. PMC 2735254 . PMID 19136955. doi:10.1038/nsmb.1545.
- Zhang, C; Darnell, RB (2011). "Mapping in vivo protein-RNA interactions at single-nucleotide resolution from HITS-CLIP data". Nature Biotechnology. 29 (7): 607–614. PMC 3400429 . PMID 21633356. doi:10.1038/nbt.1873.
- Zisoulis, DG; Lovci, MT; Wilbert, ML; Hutt, KR; Liang, TY; Pasquinelli, AE; Yeo, GW (2010). "Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans". Nat Struct Mol Biol. 17 (2): 173–179. PMC 2834287 . PMID 20062054. doi:10.1038/nsmb.1745.
- Kargapolova, Y; Levin, M; Lackner, K; Danckwardt, S (2017). "sCLIP—an integrated platform to study RNA–protein interactomes in biomedical research: identification of CSTF2tau in alternative processing of small nuclear RNAs". Nucl Acids Res. Epub ahead of print. PMID 28334977. doi:10.1093/nar/gkx152.