C9orf72
| C9orf72 | |||||||
|---|---|---|---|---|---|---|---|
| Identifiers | |||||||
| Aliases | C9orf72, chromosome 9 open reading frame 72, ALSFTD, FTDALS, FTDALS1, DENNL72 | ||||||
| External IDs | MGI: 1920455 HomoloGene: 10137 GeneCards: C9orf72 | ||||||
| Orthologs | |||||||
| Species | Human | Mouse | |||||
| Entrez | |||||||
| Ensembl | |||||||
| UniProt | |||||||
| RefSeq (mRNA) | |||||||
| RefSeq (protein) | |||||||
| Location (UCSC) | Chr 9: 27.55 – 27.57 Mb | Chr 4: 35.19 – 35.23 Mb | |||||
| PubMed search | [1] | [2] | |||||
| Wikidata | |||||||
| |||||||
C9orf72 (chromosome 9 open reading frame 72) is a protein which in humans is encoded by the gene C9orf72.
The human C9orf72 gene is located on the short (p) arm of chromosome 9 open reading frame 72, from base pair 27,546,542 to base pair 27,573,863. Its cytogenetic location is at 9p21.2.[3]
The protein is found in many regions of the brain, in the cytoplasm of neurons as well as in presynaptic terminals. Disease causing mutations in the gene were first discovered by two independent research teams, led by Rosa Rademakers of Mayo Clinic and Bryan Traynor of the National Institutes of Health, and were first reported in October 2011.[4][5] The mutations in C9orf72 are significant because it is the first pathogenic mechanism identified to be a genetic link between familial frontotemporal dementia (FTD) and of amyotrophic lateral sclerosis (ALS). As of 2012, it is the most common mutation identified that is associated with familial FTD and/or ALS.
Gene location
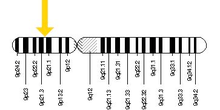
Cytogenetic Location: 9p21.2
Molecular Location on chromosome 9: base pairs 27,546,542 to 27,573,863
The C9orf72 gene is located on the short (p) arm of chromosome 9 at position 21.2.
More precisely, the C9orf72 gene is located from base pair 27,546,542 to base pair 27,573,863 on chromosome 9.[6]
Mutation
The mutation of C9ORF72 is a hexanucleotide repeat expansion of the six letter string of nucleotides GGGGCC.[7] In a normal person, there are few repeats of this hexanucleotide, typically less than 20-30,[8] but in people with the mutation, the repeat can occur in the order of hundreds.[9] It is known that the mutation interferes with normal expression of the protein made by C9orf72, however the function of that protein is unknown (though see section below). There are two major theories about the way that the C9ORF72 mutation causes FTD and/or ALS. One theory is that accumulation of RNA in the nucleus and cytoplasm becomes toxic, and RNA binding protein sequestration occurs. The other is that the lack of half of the C9ORF72 protein (Haploinsufficiency) in the body causes the diseases. Additionally, RNA transcribed from the C9ORF72 gene, containing expanded GGGGCC repeats, is translated through a non-ATG initiated mechanism. This drives the formation and accumulation of dipeptide repeat proteins corresponding multiple ribosomal reading frames on the mutation.[10][11] GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport through several possible mechanisms.[12]
Clinical significance
The C9ORF72 mutation is the first mutation found to be a link between familial FTD and ALS. Numerous published studies have confirmed the commonality of the C9ORF72 repeat expansion in FTD and ALS, which are both diseases without cures that have affected millions of people. Frontotemporal dementia is the second most common form of early-onset dementia after Alzheimer’s disease in people under the age of 65.[13] Amyotrophic lateral sclerosis is also devastating; it is characterized by motor neuron degeneration that eventually causes respiratory failure with a median survival of three years after onset.[14]
C9orf72 is present in approximately 40% of familial ALS and 8-10 % of sporadic ALS. It is currently the most common demonstrated mutation related to ALS - far more common than SOD1 or TDP-43.
While different mutations of various genes have been linked to different phenotypes of FTD in the past, C9orf72 specifically has been linked to behavioral variant FTD.[15] Certain pathology in FTD caused by the C9orf72 mutation can also include:
- TDP-43 in all C9 carriers[16]
- Ubiquitin-binding protein 62[17]
C9ORF72 is specifically linked to familial ALS, which affects about 10% of ALS patients. Traditionally, familial and sporadic cases of ALS have been clinically indistinguishable, which has made diagnosis difficult. The identification of this gene will therefore help in the future diagnosis of familial ALS.[14] Slow diagnosis is also common for FTD, which can often take up to a year with many patients initially misdiagnosed with another condition. Testing for a specific gene that is known to cause the diseases would help with faster diagnoses. Possibly most importantly, the identification of this hexanucleotide repeat expansion is an extremely promising avenue for possible future therapies of both familial FTD and familial ALS, once the mechanism and function of the C9ORF72 protein is better comprehended. Furthermore, present research is being done to see if there is a correlation between C9ORF72 and other neurological diseases, such as motor neuron disease and Huntington's disease.[18][19]
Gene heritability
It is possible that genetic anticipation may exist for this mutation, However, only 1 in 4 families exhibited significant anticipation in this study (n=63) [16] It has been proposed that the amount of the repeat expansion increases with each successive generation, possibly causing the disease to be more severe in the next generation, showing onset up to a decade earlier with each successive generation after the carrier. The buildup of a repeat expansion with each generation is typically thought to occur because the DNA is unstable and therefore accumulates exponentially every time the gene is copied. No genetic evidence for this has yet been demonstrated for this mutation.[20] There is also a demographic factor that should be considered in genetic predisposition, as some cohorts have found that there might be a founder effect for the C9orf72 mutation, which might have led to higher frequencies of the mutation in specific populations than others. Specifically this founder has been linked to Northern Europeans populations, namely Finland.[15]
Gene testing
Since this mutation has been found to be the most common mutation identified in familial FTD and/or ALS, it is considered one of if not the most dependable candidates for genetic testing. Patients are considered eligible if the mother or father has had FTD and/or another family member has had ALS.[14] There are also population and location risk factors in determining eligibility. Some studies have found that the mutation has a higher frequency in certain cohorts.[21] Athena Diagnostics (Quest Diagnostics) announced in Spring 2012 the first clinically available testing service for detecting the hexanucleotide repeat expansion in the C9orf72 gene.[22] Genetic counseling is recommended for the patients before a genetic test is ordered.
Likely function
C9ORF72 is a full-length homologue of DENN proteins (where DENN stands for "differentially expressed in normal and neoplastic cells").[23][24][25] These proteins have a conserved DENN module consisting of an N-terminal longin domain, followed by the central DENN and C-terminal alpha-helical d-DENN domains.[24] This has led to DENNL72 being suggested as a new name for C9orf72.[25]
Given the molecular role of known DENN modules,[26] the C9ORF72-like proteins are predicted to function as Guanine nucleotide exchange factors for small GTPases, most likely a Rab. A recent study provided the first experimental evidence to confirm this: C9ORF72 was found to regulate endosomal trafficking and autophagy in neuronal cells and primary neurons.[24][27] This suggests that certain aspects of the ALS and FTD disease pathology might result from haploinsufficiency of C9ORF72/DENNL72, leading to a defect in intracellular membrane traffic, either exocytosis or endocytosis, in addition to the strong possibility of RNA-mediated toxicity.
Evolutionary history
Sequence analysis further suggests that the C9ORF72 protein emerged early in eukaryotic evolution, and whereas most eukaryotes usually possess a single copy of the gene encoding the C9ORF72 protein, the eukaryotes Entamoeba and Trichomonas vaginalis possess multiple copies, suggestive of independent lineage-specific expansions in these species. Interestingly, the family is lost in most fungi (except Rhizopus) and plants.[24][25]
Implications for future therapies
Overall, the C9ORF72 mutation holds great promise for future therapies for familial FTD and/or ALS to be developed. Currently, there is focus on more research to be done on C9ORF72 to further understand the exact mechanisms involved in the cause of the diseases by this mutation. A clearer understanding of the exact pathogenic mechanism will aid in a more focused drug therapies. Possible drug targets currently include the repeat expansion itself as well as increasing levels of C9ORF72. Blocking the toxic gain of RNA foci to prevent RNA sequestration might be helpful as well as making up for the lack of C9ORF72. Either of these targets as well as a combination of them might be promising future targets in minimizing the effects of the C9ORF72 repeat expansion.[28]
Interactions
C9ORF72 has been shown to interact with:
References
- ↑ "Human PubMed Reference:".
- ↑ "Mouse PubMed Reference:".
- ↑ C9orf72 chromosome 9 open reading frame 72 [Homo sapiens] - Gene - NCBI
- ↑ DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R (2011). "Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS". Neuron. 72 (2): 245–56. PMC 3202986
 . PMID 21944778. doi:10.1016/j.neuron.2011.09.011.
. PMID 21944778. doi:10.1016/j.neuron.2011.09.011. - ↑ Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Hölttä-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chiò A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ (2011). "A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD". Neuron. 72 (2): 257–68. PMC 3200438 . PMID 21944779. doi:10.1016/j.neuron.2011.09.010.
- ↑ "C9orf72". Retrieved July 2013. Check date values in:
|access-date=(help) - ↑ Bigio EH (2011). "C9ORF72, the new gene on the block, causes C9FTD/ALS: new insights provided by neuropathology". Acta Neuropathol. 122 (6): 653–5. PMC 3262229 . PMID 22101324. doi:10.1007/s00401-011-0919-7.
- ↑ Fong JC, Karydas AM, Goldman JS (2012). "Genetic counseling for FTD/ALS caused by the C9ORF72 hexanucleotide expansion". Alzheimers Res Ther. 4 (4): 27. PMC 3506941 . PMID 22808918. doi:10.1186/alzrt130.
- ↑ Khan BK, Yokoyama JS, Takada LT, Sha SJ, Rutherford NJ, Fong JC, Karydas AM, Wu T, Ketelle RS, Baker MC, Hernandez MD, Coppola G, Geschwind DH, Rademakers R, Lee SE, Rosen HJ, Rabinovici GD, Seeley WW, Rankin KP, Boxer AL, Miller BL (2012). "Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion". J. Neurol. Neurosurg. Psychiatr. 83 (4): 358–64. PMC 3388906 . PMID 22399793. doi:10.1136/jnnp-2011-301883.
- ↑ Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D (2013). "The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS". Science. 339 (6125): 1335–8. PMID 23393093. doi:10.1126/science.1232927.
- ↑ Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, Rademakers R, Boylan KB, Dickson DW, Petrucelli L (2013). "Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS". Neuron. 77 (4): 639–46. PMC 3593233 . PMID 23415312. doi:10.1016/j.neuron.2013.02.004.
- ↑ Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP (2015). "GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport". Nature. 525: 129–33. PMC 4631399 . PMID 26308899. doi:10.1038/nature14974.
- ↑ Ratnavalli E, Brayne C, Dawson K, Hodges JR (2002). "The prevalence of frontotemporal dementia". Neurology. 58 (11): 1615–21. PMID 12058088. doi:10.1212/WNL.58.11.1615.
- 1 2 3 Herdewyn S, Zhao H, Moisse M, Race V, Matthijs G, Reumers J, Kusters B, Schelhaas HJ, van den Berg LH, Goris A, Robberecht W, Lambrechts D, Van Damme P (2012). "Whole-genome sequencing reveals a coding non-pathogenic variant tagging a non-coding pathogenic hexanucleotide repeat expansion in C9orf72 as cause of amyotrophic lateral sclerosis". Hum. Mol. Genet. 21 (11): 2412–9. PMC 3349421 . PMID 22343411. doi:10.1093/hmg/dds055.
- 1 2 Friedland RP, Shah JJ, Farrer LA, Vardarajan B, Rebolledo-Mendez JD, Mok K, Hardy J (2012). "Behavioral variant frontotemporal lobar degeneration with amyotrophic lateral sclerosis with a chromosome 9p21 hexanucleotide repeat". Front Neurol. 3: 136. PMC 3463813 . PMID 23060854. doi:10.3389/fneur.2012.00136.
- 1 2 Boeve BF, Boylan KB, Graff-Radford NR, DeJesus-Hernandez M, Knopman DS, Pedraza O, Vemuri P, Jones D, Lowe V, Murray ME, Dickson DW, Josephs KA, Rush BK, Machulda MM, Fields JA, Ferman TJ, Baker M, Rutherford NJ, Adamson J, Wszolek ZK, Adeli A, Savica R, Boot B, Kuntz KM, Gavrilova R, Reeves A, Whitwell J, Kantarci K, Jack CR, Parisi JE, Lucas JA, Petersen RC, Rademakers R (2012). "Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72". Brain. 135 (Pt 3): 765–83. PMC 3286335 . PMID 22366793. doi:10.1093/brain/aws004.
- ↑ Mahoney CJ, Beck J, Rohrer JD, Lashley T, Mok K, Shakespeare T, Yeatman T, Warrington EK, Schott JM, Fox NC, Rossor MN, Hardy J, Collinge J, Revesz T, Mead S, Warren JD (2012). "Frontotemporal dementia with the C9ORF72 hexanucleotide repeat expansion: clinical, neuroanatomical and neuropathological features". Brain. 135 (Pt 3): 736–50. PMC 3286330 . PMID 22366791. doi:10.1093/brain/awr361.
- ↑ Otomo A, Pan L, Hadano S (2012). "Dysregulation of the autophagy-endolysosomal system in amyotrophic lateral sclerosis and related motor neuron diseases". Neurol Res Int. 2012: 498428. PMC 3407648 . PMID 22852081. doi:10.1155/2012/498428.
- ↑ Hensman Moss DJ, Poulter M, Beck J, Hehir J, Polke JM, Campbell T, Adamson G, Mudanohwo E, McColgan P, Haworth A, Wild EJ, Sweeney MG, Houlden H, Mead S, Tabrizi SJ (2014). "C9orf72 expansions are the most common genetic cause of Huntington disease phenocopies". Neurology. 82 (4): 292–9. PMC 3929197 . PMID 24363131. doi:10.1212/WNL.0000000000000061.
- ↑ http://www.ncbi.nlm.nih.gov/books/NBK268647/
- ↑ Sieben A, Van Langenhove T, Engelborghs S, Martin JJ, Boon P, Cras P, De Deyn PP, Santens P, Van Broeckhoven C, Cruts M (2012). "The genetics and neuropathology of frontotemporal lobar degeneration". Acta Neuropathol. 124 (3): 353–72. PMC 3422616 . PMID 22890575. doi:10.1007/s00401-012-1029-x.
- ↑ New Testing for ALS (2012)
- ↑ Söding J, Biegert A, Lupas AN (2005). "The HHpred interactive server for protein homology detection and structure prediction". Nucleic Acids Res. 33 (Web Server issue): W244–8. PMC 1160169 . PMID 15980461. doi:10.1093/nar/gki408.
- 1 2 3 4 Zhang D, Iyer LM, He F, Aravind L (2012). "Discovery of Novel DENN Proteins: Implications for the Evolution of Eukaryotic Intracellular Membrane Structures and Human Disease". Front Genet. 3: 283. PMC 3521125 . PMID 23248642. doi:10.3389/fgene.2012.00283.
- 1 2 3 Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ (2013). "The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs". Bioinformatics. 29 (4): 499–503. PMC 3570213 . PMID 23329412. doi:10.1093/bioinformatics/bts725.
- ↑ Yoshimura S, Gerondopoulos A, Linford A, Rigden DJ, Barr FA (2010). "Family-wide characterization of the DENN domain Rab GDP-GTP exchange factors". J. Cell Biol. 191 (2): 367–81. PMC 2958468 . PMID 20937701. doi:10.1083/jcb.201008051.
- ↑ Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD (2014). "C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking". Human Molecular Genetics. 23: 3579–3595. PMC 4049310 . PMID 24549040. doi:10.1093/hmg/ddu068.
- ↑ Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, Rutherford NJ, Baker M, Knopman DS, Wszolek ZK, Parisi JE, Dickson DW, Petersen RC, Rademakers R, Jack CR, Josephs KA (2012). "Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics". Brain. 135 (Pt 3): 794–806. PMC 3286334 . PMID 22366795. doi:10.1093/brain/aws001.
- 1 2 "C9orf72 Interaction Summary". BioGRID.
- ↑ Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Heusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD (2013). "RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention". Neuron. 80 (2): 415–28. PMC 4098943 . PMID 24139042. doi:10.1016/j.neuron.2013.10.015.
External links
- Human C9orf72 genome location and C9orf72 gene details page in the UCSC Genome Browser.
| Wikimedia Commons has media related to C9orf72. |