Hereditary angioedema
| Hereditary angioedema | |
|---|---|
| Synonyms | Hereditary angioneurotic edema (HANE),[1] familial angioneurotic edema[2] |
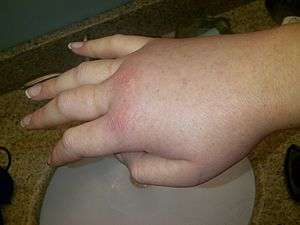 | |
| Swollen right hand during a hereditary angioedema attack. | |
| Specialty | Hematology |
| Symptoms | Recurrent attacks of severe swelling[3] |
| Usual onset | Childhood[3] |
| Duration | Attacks last a few days[3] |
| Types | Type I, II, III[3] |
| Causes | Genetic disorder (autosomal dominant)[3] |
| Diagnostic method | Measuring C4 and C1-inhibitor levels.[2] |
| Similar conditions | Intestinal obstruction, other types of angioedema[2] |
| Prevention | C1 esterase inhibitor[1] |
| Treatment | Supportive care, medications[1] |
| Medication | C1 esterase inhibitor, ecallantide, icatibant[1] |
| Prognosis | 25% risk of death if airway involved (without treatment)[2] |
| Frequency | ~1 in 50,000[3] |
Hereditary angioedema (HAE) is disorder that results in recurrent attacks of severe swelling.[3] This most commonly affects the arms, legs, face, intestinal tract, and airway.[3] Itchiness does not typically occur.[2] If the intestinal tract is affected abdominal pain and vomiting may occur.[1] Swelling of the airway can result in its obstruction.[1] Attacks, without treatment, typically occur every couple of weeks and last for a few days.[3]
There are three main types of HAE.[3] Type I and II are caused by a mutation in the SERPING1 gene that makes the C1 inhibitor protein while type III is often due to a mutation of the factor XII gene.[3] This results in increased amounts of bradykinin which promotes swelling.[3] The condition may be inherited from a person's parents in an autosomal dominant manner or occur as a new mutation.[3] Triggers of an attack may include minor trauma or stress, but often occurs without any obvious preceding event.[3] Diagnosis of type I and II is based upon measuring C4 and C1-inhibitor levels.[2]
Management involves efforts to prevent attacks and the treatment of attacks if they occur.[1] During an attack supportive care such as intravenous fluids and airway support may be required.[1] The medication C1 esterase inhibitor can be used for both prevention and treatment.[1] Ecallantide and icatibant can be used to treat acute attacks.[1]
This disorder affects approximately one in 50,000 people.[3] The condition is typically first noticed in childhood.[3] Type I and II affected females and males equally.[4] Type III affects females more often than males.[2] When the airway is involved, without treatment, death occurs in about 25%.[2] With treatment outcomes are generally good.[2] The condition was first described in 1888 by William Osler.[5]
Signs and symptoms
Symptoms generally begin around puberty but can occur earlier. These individuals have recurrent swelling in the extremities, genitals, face, lips, larynx or GI tract. Some patients describe a sensation of fullness but not pain or itching in the affected area except for those with abdominal swellings who often experience acute abdominal pain. Others experience an intense amount of pain, described as radiating from the bone outward along with intense itching just beneath the skin and intense heat, regardless of the area targeted.
Instances of swelling around the throat or larynx can cause difficulties in breathing should the swelling obstruct airways. This has been known to cause a large number of fatalities in those afflicted with the disorder. Episodes that attack the gastrointestinal tract can cause a number of complications including dehydration from being unable to keep anything down (which, depending on length of the episode, can prove fatal). Symptoms from gI tract swelling including violent vomiting, intense pain from the midsection, dehydration, and intense exhaustion.
Some suffered of HAE suffer from 'wandering' attacks. These attacks will center around an extremity. For example: Should the sufferer's hand swell up, it will go through the normal swelling cycle before 'transferring' to either the connection limb (In this case wrist to forearm) or move to the opposite hand. Sufferers with this symptom may find their episodes last longer, and may find their triggers more difficult to track.
Pathogenesis
Because hereditary angioedema is an autosomal inheritable disease, there is no gender difference in transmission and both sexes are equally likely to receive the mutated gene from their parent(s). The figure below [What figure?] (courtesy of US National Library Of Medicine) depicts autosomal dominant transmission. Here, the father (individual A) with a mutated gene for HAE, has the disease while his wife (individual B) with 2 non-mutated copies of the C1 inhibitor gene and does not have the disease. The possibility of a cross between them gives the possibilities as shown: two of their offspring will have the disease (HEA) while the others would not.[6][7]
The affected father who has HAE has a mutation on one of his genes (C1-INH). Each one of his children, notwithstanding his/her sex, will have a 50% chance to inherit the mutated C1-INH gene from him. HAE is generally referred to as a "dominant" condition because it only takes a mutation in one of the two C1-INH genes in a carrier to cause the disease.[8][9]
The prevalence of HAE is relatively low – between 1 in every 10,000 to 1 in every 50,000 persons. Most persons with HAE acquire a C1 esterase inhibitor (C1-INH) mutation from one of their parents. A parent with HAE usually has a 50% probability of transmitting this condition on to one of his/her children of either sex as shown in the figure (HEA Inheritance). In occasions when HAE is not inherited and occurs in people with no previous history of it. This is because there are new impulsive or spontaneous changes in the sperm or egg cell that is responsible for this specific pregnancy. In a review of patients who do not have a history of HAE in their family, but who have relatively low levels of mutated C1-INH with persistent angioedema, 25% of new patients who had HAE had C1-INH changes that do not show signs of being inherited but rather new.
The mutational changes in 1 or both of the carriers' C1 inhibitor genes could have only occurred spontaneously, and just like in the example above, their offspring in this case will have a 50% probability of acquiring the mutated gene from either parent that has HAE.
Diagnosis
| C4 (C) | FB (A) | C3 | CH50 | Conditions |
|---|---|---|---|---|
| · | ↓ | ↓ | ↓ | PSG, C3 NeF AA |
| ↓ | · | ↓ | · | HA, C4D |
| · | · | · | ↓ | TCPD |
| ↓ | · /↓ | ↓ | ↓ | SLE |
| ↑ | ↑ | ↑ | ↑ | inflammation |
Recognizing HAE is often difficult due to the wide variability in disease expression. The course of the disease is diverse and unpredictable, even within a single patient over their lifetime. This disease may be similar in its presentation to other forms of angioedema resulting from allergies or other medical conditions, but it is significantly different in cause and treatment. When hereditary angioedema is misdiagnosed as an allergy it is most commonly treated with steroids and epinephrine, drugs that are usually ineffective in treating a hereditary angioedema episode. Other misdiagnoses have resulted in unnecessary exploratory surgery for patients with abdominal swelling and other hereditary angioedema patients report that their abdominal pain was wrongly diagnosed as psychosomatic.
HAE accounts for only a small fraction of all cases of angioedema. To avoid potentially fatal consequences such as upper airway obstruction and unnecessary abdominal surgery, the importance of a correct diagnosis cannot be over-emphasized.[10]
Consider hereditary angioedema (HAE) if a patient presents with:
- Recurrent angioedema (without urticaria)
- Recurrent episodes of abdominal pain and vomiting
- Laryngeal edema
- Positive family history of angioedema
[11][12] A blood test, ideally taken during an episode, can be used to diagnose the condition. Measure: serum complement factor 4 (C4), C1 inhibitor (C1-INH) antigenic protein, C1 inhibitor (C1-INH) functional level if available.Analysis of complement C1 inhibitor levels may play a role in diagnosis. C4 and C2 are complementary components.
Types
There are three types of C1 inhibitor deficiency:[13]
HAE type I is primarily caused by a deficiency in blood proteins (C1 esterase inhibitors) which normally suppress activation of the complement system. The resultant over-stimulation of this system leads to the production of inflammatory anaphylatoxins, which affects the flow of body fluids between the vascular system and body tissues. This deficiency is responsible for approximately 80-85% of cases.
HAE type II is a less frequently encountered form of this disorder and accounts for 15-20% of cases. In this type, atypical C1-inhibitor proteins are produced which are less capable of suppressing activation of the complement system. Like HAE type I, this results in over-stimulation of this system.
HAE type III is rare and has only been documented recently. Unlike types I and II, this form does not appear to be connected with C1-inhibitor deficiency. This type mainly affects females and appears to be influenced by contact with estrogens and also by hormone replacement therapy (e.g. oral contraceptives). Its pathogenesis is credited to increased activity of the enzyme kininogenase, which leads to rise in the levels of bradykinin. Other patients with type III HAE have alterations in gene F12, which encodes a protein which participates in blood coagulation.[14] Some patients with type III HAE have a mutation in the F12 gene which produces a protein involved in blood clotting.[15]
Prevention
Treatment with ACE inhibitors is contraindicated in this condition, as these drugs can lead to bradykinin accumulation, which can precipitate disease episodes.[16][17]
Long-term
Patients in whom episodes occur at least once a month or who are at high risk of developing laryngeal edema require long-term prevention. There are now several phase III clinical trials recently published in HAE prophylaxis and therapy and these have led to the licensing of pdC1INH (Berinert®, CSL Behring; Cinryze®, ViroPharma; Cetor-n®, Sanquin) in many parts of the world; bradykinin receptor antagonist (Icatibant, Firazyr®, Jerini/Shire) in Europe; kallikrein inhibitor(Ecallantide, Kalbitor®, Dyax) in the United States; and recombinant C1-INH replacement therapy (rhC1INH; conestat alfa; Rhucin®, Pharming) in Europe. Tranexamic acid has been shown to be relatively ineffective therapy. Danazol prophylaxis remains an option but therapeutic agents are now being used more for prophylaxis because of danazol adverse events.[18] For patients requiring long-term prophylaxis, home therapy which allows patients to self-administer product, is considered an integral part of allowing patients a normal quality of life.
Short-term
Short-term prevention is normally administered before surgery or dental treatment. In Germany, C1-INH concentrate is used for this and given 1–1.5 hours before the procedure. In countries where C1-inhibitor concentrate is not available or only available in an emergency (laryngeal edema), high-dose androgen treatment is administered for 5–7 days.
Management
The aim of acute treatment is to halt progression of the edema as quickly as possible, which can be life-saving, particularly if the swelling is in the larynx. In Germany, most acute treatment consists of C1 inhibitor concentrate from donor blood, which must be administered intravenously; however, in most European countries, C1 inhibitor concentrate is only available to patients who are participating in special programs. In emergency situations where C1 inhibitor concentrate is not available, fresh frozen plasma (FFP) can be used as an alternative, as it also contains C1 inhibitor.
Other treatment modalities can stimulate the synthesis of C1 inhibitor, or reduce C1 inhibitor consumption. Purified C1 inhibitor, derived from human blood, has been used in Europe since 1979. Several C1 inhibitor treatments are now available in the U.S. Food and Drug Administration and two C1 inhibitor products are now available in Canada. Berinert P (CSL Behring), which is pasteurized, was approved by the F.D.A. in 2009 for acute attacks. Cinryze (ViroPharma), which is nanofiltered, was approved by the F.D.A. in 2008 for prophylaxis. Ruconest (Pharming) is a recombinant C1 inhibitor approved in the US and Europe that does not carry the risk of infectious disease transmission due to human blood-borne pathogens.[19]
The medication ecallantide inhibits plasma kallikrein, and was approved by the F.D.A. (but not in Europe) for acute attacks in 2009. Icatibant inhibits the bradykinin B2 receptor, and was approved in Europe and the USA.[19][20] In hereditary angioedema, specific stimuli that have previously led to attacks may need to be avoided in the future. It does not respond to antihistamines, corticosteroids, or epinephrine.
Prognosis
A 2014 review stated that 25% and 30% of identified suffers die in the first two decades of life, mainly due to lack of treatment.[21]
Epidemiology
Data regarding the epidemiology of angioedema is limited. The incidence of HAE is one in 10,000–50,000 people in the United States and Canada. Mortality rates are estimated at 15–33%, resulting primarily from laryngeal edema and asphyxiation. HAE leads to 15,000–30,000 emergency department visits per year.[22][23]
Patients' organizations
There are national associations for HAE patients and their families in a number of countries around the world. These national associations are members of the global organization HAEi - International Patient Organization for C1-Inhibitor Deficiencies. HAEi is dedicated to raising awareness of C1 inhibitor deficiencies around the world. It is a non-profit international network established to promote co-operation, co-ordination and information sharing between HAE specialists and national HAE patient associations in order to help facilitate the availability of effective diagnosis and management of C1 inhibitor deficiencies throughout the world.[24]
The Assistance Fund Inc. is an American nonprofit organization that offers co-pay assistance for medications that treat HAE and is open to any American Citizens or landed immigrants who have insurance.
Research
Clinical development of several new active substances, which intervene in the disease process in different ways, is currently ongoing.
Pharming Group NV announced on 24 June 2010 that the European Medicines Agency has adopted a positive opinion on conestat alfa (trade name Ruconest), a C1-inhibitor for the treatment of acute angioedema attacks.[25]
Ecallantide, a peptide inhibitor of kallikrein, has received orphan status for HAE and has shown positive results in phase III trials.[26]
Icatibant (marketed as Firazyr) is a selective bradykinin receptor antagonist, which has been approved in Europe and was approved in the USA by the FDA in Aug 2011.[27] After initial borderline results this drug was shown to be effective in phase III trials.[28] Cinryze has been approved by the FDA in October 2008.[29]
References
- 1 2 3 4 5 6 7 8 9 10 "Hereditary angioedema". GARD. 2017. Retrieved 10 July 2017.
- 1 2 3 4 5 6 7 8 9 "Orphanet: Hereditary angioedema". www.orpha.net. August 2011. Retrieved 10 July 2017.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 Reference, Genetics Home (5 July 2017). "hereditary angioedema". Genetics Home Reference. Retrieved 10 July 2017.
- ↑ "Hereditary Angioedema - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). 2008. Retrieved 10 July 2017.
- ↑ Levin, Alex V.; Enzenauer, Robert W. (2017). The Eye in Pediatric Systemic Disease. Springer. p. 71. ISBN 9783319183893.
- ↑ Ferraro, M. F.; Moreno, A. S.; Castelli, E. C.; Donadi, E. A.; Palma, M. S.; Arcuri, H. A.; Lange, A. P.; Bork, K.; Sarti, W.; Arruda, L. K. Allergy. Oct2011, Vol. 66 Issue 10, p1384-1390. 7p. 2 Diagrams, 1 Chart. DOI: 10.1111/j.1398-9995.2011.02658.x.
- ↑ Bafunno, Valeria; Bova, Maria; Loffredo, Stefania; Divella, Chiara; Petraroli, Angelica; Marone, Gianni; Montinaro, Vincenzo; Margaglione, Maurizio; Triggiani, Massimo. Annals of Human Genetics. Mar2014, Vol. 78 Issue 2, p73-82. 10p. DOI: 10.1111/ahg.12052.
- ↑ [Genetic test indications and interpretations in patients with hereditary angioedema. Weiler CR, van Dellen RG. Mayo Clin Proc. 2006 Jul;81(7):958-72. Review.]
- ↑
- ↑ http://caep.ca/sites/caep.ca/files/caep/files/2012_conference_-_management_of_angioedema_in_the_er.pdf
- ↑ Zingale LC, Beltrami L, Zanichelli A, et al. (October 2006). "Angioedema without urticaria: a large clinical survey". CMAJ. 175 (9): 1065–70. PMC 1609157
 . PMID 17060655. doi:10.1503/cmaj.060535.
. PMID 17060655. doi:10.1503/cmaj.060535. - ↑ "Diagnostic Algorithm". HAE Canada.
- ↑ Gompels MM, Lock RJ, Abinun M, Bethune CA, Davies G, Grattan C, Fay AC, Longhurst HJ, Morrison L, Price A, Price M, Watters D (March 2005). "C1 inhibitor deficiency: consensus document" (pdf). Clinical and Experimental Immunology. 139 (3): 379–94. PMC 1809312 . PMID 15730382. doi:10.1111/j.1365-2249.2005.02726.x.
- ↑ "More Information". HAE UK. Archived from the original on 2014-04-22.
- ↑ [Type III hereditary angioedema: defined, but not understood. Kaplan A. Ann Allergy Asthma Immunol. 2012 Sep;109(3):153-4. doi: 10.1016/j.anai.2012.07.007. No abstract available.]
- ↑ Dendorfer A, Wolfrum S, Wagemann M, Qadri F, Dominiak P (May 2001). "Pathways of bradykinin degradation in blood and plasma of normotensive and hypertensive rats". Am. J. Physiol. Heart Circ. Physiol. 280 (5): H2182–8. PMID 11299220.
- ↑ Kuoppala A, Lindstedt KA, Saarinen J, Kovanen PT, Kokkonen JO (April 2000). "Inactivation of bradykinin by angiotensin-converting enzyme and by carboxypeptidase N in human plasma". Am. J. Physiol. Heart Circ. Physiol. 278 (4): H1069–74. PMID 10749699.
- ↑ Hereditary angioedema: beyond international consensus - circa December 2010 - The Canadian Society of Allergy and Clinical Immunology Dr. David McCourtie Lecture
- 1 2 Morgan
- ↑ Firazyr [package insert]. Lexington, MA: Shire Orphan Therapies, Inc; 2011.
- ↑ Varga, Lilian; Farkas, Henriette (2008-11-01). "Treatment of type I and II hereditary angioedema with Rhucin®, a recombinant human C1 inhibitor". Expert Review of Clinical Immunology. 4 (6): 653–661. ISSN 1744-666X. doi:10.1586/1744666X.4.6.653.
- ↑ From the: Pinnacle Health System, Harrisburg Hospital, Department of Internal Medicine, 111 South Front Street, Harrisburg, PA 17101, Update on treatment for her
- ↑ "Update on treatment of hereditary angioedema" Buyantseva, Larisa, Sardana, Niti and Craig, Timothy
- ↑ "HAEi website".
- ↑ Pharming: Pharming Receives Positive Opinion From European Medicines Agency On Rhucin Product name in Europe changed to Ruconest
- ↑ Lehmann A (August 2008). "Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery". Expert Opin Biol Ther. 8 (8): 1187–99. PMID 18613770. doi:10.1517/14712598.8.8.1187.
- ↑ Jerini AG (2008-07-15). "Jerini Receives European Commission Approval for Firazyr (Icatibant) in the Treatment of HAE - Press release". Retrieved 2008-07-28.
- ↑ Bernstein JA (January 2008). "Hereditary angioedema: a current state-of-the-art review, VIII: current status of emerging therapies". Ann. Allergy Asthma Immunol. 100 (1 Suppl 2): S41–6. PMID 18220151. doi:10.1016/S1081-1206(10)60585-6.
- ↑ Reuters: UPDATE 1-US clears Lev Pharma drug for rare swelling disease
{kind=link}
External links
| Classification |
V · T · D |
|---|---|
| External resources |