Brook rearrangement
The Brook rearrangement in organic chemistry is a rearrangement reaction in which an organosilyl group switches position with a hydroxyl proton over a carbon to oxygen covalent bond under the influence of a base.[1] It is named for the Canadian chemist Adrian Gibbs Brook (1924–2013). The reaction product is a silyl ether.
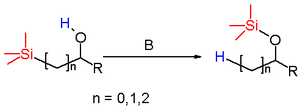
The silyl substituents can be aliphatic (methyl) or aromatic (phenyl) and the alcohol is secondary or tertiary with aliphatic or aryl groups. The base is an amine, sodium hydroxide, an organolithium reagent or an alkali metal alloy such as sodium/potassium. When the reactant is a silylmethanol the reaction is a 1,2-brook rearrangement but rearrangements over larger carbon skeletons are also possible.
Reaction mechanism
The reaction mechanism for this rearrangement starts with proton abstraction of the hydroxyl group by base to the alkoxide anion. This nucleophile attacks the silicon atom in a nucleophilic displacement with the methylene group as the leaving group. The proposed transition state for this reaction step is a three-membered ring with the degree of Si-O bond making in step with the Si-C bond breaking process. The additional electron pair is now transferred from oxygen to a carbanion which quickly abstracts a proton from a proton source such as solvent to form the final silyl ether.
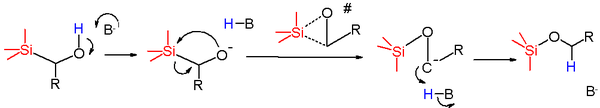
When the reactant is (triphenylsilyl)methylphenylmethanol the activation energy is found to be relatively low but that the entropy of activation has a large negative value which supports the cyclic transition state structure. The Hammett results for a group of para-substituted phenyl methanols conform that electron withdrawing groups help to stabilize the negative charge built up in the carbanionic intermediate.
The thermodynamic driving force for this reaction is the formation of a silicon oxygen bond. The cleavage of the Si-C bond and O-H bond costs in terms of bond dissociation energy are 451 + 427 = 878 kJ/mol and the gains are 809 (Si-O) + 338 (C-H) = 1147 kJ/mol.
The Brook rearrangement occurs with retention of configuration as demonstrated in a Walden cycle:
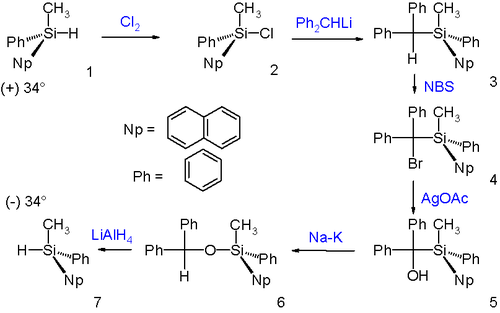
The (+)- silyl hydride enantiomer reacts with chlorine to the corresponding silyl chloride with retention of the stereocenter. The subsequent nucleophilic displacement of chlorine with diphenylmethyl lithium occurs with inversion. The next two steps convert the diphenylmethine group through bromination with NBS (or N-bromosuccinimide) and hydrolysis with silver acetate into a diphenylmethanol group without any change in configuration. The subsequent Brook rearrangement to the silyl ether and reduction with lithium aluminium hydride occur with retention so that the final reaction product is the opposite enantiomer of the starting material with opposite sign for specific rotation.
Scope
Brook rearrangements are known in acylsilanes [2] and with special silyl ethers retro-brook rearrangements are also possible.[3][4] An analogous phospha-Brook rearrangement of 1-hydroxyphosphonates has also been reported.
References
- ↑ A. G. Brook (1974). "Molecular rearrangements of organosilicon compounds". Acc. Chem. Res. 7 (3): 77–84. doi:10.1021/ar50075a003.
- ↑ Patrocinio, Amauri F. and Moran, Paulo J. S. Acylsilanes and their applications in organic chemistry. J. Braz. Chem. Soc., 2001, vol.12, no.1, p.07-31. ISSN 0103-5053. Online article
- ↑ Stereochemistry of the cyclization of 4-(t-butyldimethyl)siloxy-5-hexenyllithium: cis-selective ring-closure accompanied by retro-[1,4]-Brook rearrangement William F. Bailey and Xinglong Jiang Arkivoc 2005 (vi) 25-32 Online article
- ↑ Higashiya, Seiichiro; Chung, Woo Jin; Lim, Dong Sung; Ngo, Silvana C.; Kelly, William H. IV; Toscano, Paul J.; Welch, John T. (2004). "Synthesis of Mono- and Difluoroacetyltrialkylsilanes and the Corresponding Enol Silyl Ethers". J. Org. Chem. 69 (19): 6323–6328. doi:10.1021/jo049551o. Retrieved 13 July 2015.