Alkaptonuria
| Alkaptonuria | |
|---|---|
 | |
| Pigmentation of the face in alkaptonuria | |
| Classification and external resources | |
| Specialty | endocrinology |
| ICD-10 | E70.2 (ILDS E70.210) |
| ICD-9-CM | 270.2 |
| OMIM | 203500 |
| DiseasesDB | 409 |
| MedlinePlus | 001200 |
| eMedicine | ped/64 |
| Patient UK | Alkaptonuria |
| MeSH | D000474 |
| GeneReviews | |
Alkaptonuria (black urine disease, black bone disease, or alcaptonuria) is a rare inherited genetic disorder in which the body cannot process the amino acids phenylalanine and tyrosine, which occur in protein. It is caused by a mutation in the HGD gene for the enzyme homogentisate 1,2-dioxygenase (EC 1.13.11.5); if a person inherits abnormal copies from each parent (it is a recessive condition) the body accumulates an intermediate substance called homogentisic acid in the blood and tissues. Homogentisic acid and its oxidized form alkapton are excreted in the urine, giving it an unusually dark color. The accumulating homogentisic acid causes damage to cartilage (ochronosis, leading to osteoarthritis) and heart valves as well as precipitating as kidney stones and stones in other organs. Symptoms usually develop in people over thirty years old, although the dark discoloration of the urine is present from birth.
Apart from treatment of the complications (such as pain relief and joint replacement for the cartilage damage), vitamin C has been used to reduce the ochronosis and lowering of the homogentisic acid levels may be attempted with a low-protein diet. It is not proven that Vitamin C works and has been since discredited. Recently the drug nitisinone has been found to suppress homogentisic acid production, and research is ongoing as to whether it can improve symptoms. Alkaptonuria is a rare disease; it occurs in one in 250,000 people, but is more common in Slovakia and the Dominican Republic.
Signs and symptoms

Patients with black bone disease are asymptomatic as children or young adults, but their urine may turn brown or even inky black if collected and left exposed to open air.[1] Pigmentation may be noted in the cartilage of the ear as well as other cartilage,[1][2] and the sclera and corneal limbus of the eye.[3]
After the age of thirty people begin to develop pain in the weight-bearing joints of the spine, hips and knees. The pain can be severe to the point that interferes with activities of daily living and may affect ability to work. Joint replacement surgery (hip and shoulder) is often necessary at a relatively young age.[1] In the longer term, the involvement of the spinal joints leads to reduced movement of the rib cage and can affect breathing.[1] Bone mineral density may be affected, increasing the risk of bone fractures, and rupture of tendons and muscles may occur.[1]
Valvular heart disease, mainly calcification and regurgitation of the aortic and mitral valves, may occur, and in severe and progressive cases valve replacement may be necessary. Irregularities in the heart rhythm and heart failure affect a significant proportion of people with alkaptonuria (40% and 10% respectively).[1] Hearing loss affects 40% of people. There is also a propensity to developing kidney stones, and eventually also gallstones and stones in the prostate and salivary glands (sialolithiasis).[1]
Pathophysiology
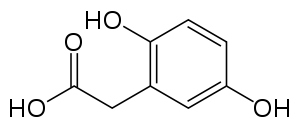
Every person carries in their DNA two copies (one received from each parent) of the gene HGD, which contains the genetic information to produce the enzyme homogentisate 1,2-dioxygenase (HGD) which can normally be found in numerous tissues in the body (liver, kidney, small intestine, colon and prostate). In people with alkaptonuria, both copies of the gene contain abnormalities that mean that the body cannot produce an adequately functioning enzyme.[4] HGD mutations are generally found in certain parts (exons 6, 8, 10 and 13) but a total of over 100 abnormalities have been described throughout the gene.[4] The normal HGD enzyme is a hexamer (it has six subunits) that are organized in two groups of three (two trimers) and contains an iron atom. Different mutations may affect the structure, function or solubility of the enzyme.[4] Very occasionally the disease appears to be transmitted in an autosomal dominant fashion, where a single abnormal copy of HGD from a single parent is associated with alkaptonuria; it is possible that other mechanisms or defects in other genes are responsible in those cases.[4]

The HGD enzyme in involved in the metabolism (chemical processing) of the aromatic amino acids phenylalanine and tyrosine. Normally these enter the bloodstream through protein-containing food and the natural turnover of protein in the body. Tyrosine is specifically required for a number of functions such as hormones (e.g. thyroxine, the thyroid hormone), melanin (the dark pigment in the skin and hair) and certain proteins, but the vast majority (over 95%) is unused and is metabolized through a group of enzymes that eventually generate acetoacetate and malate.[1] In alkaptonuria the HGD enzyme cannot metabolize the homogentisic acid (generated from tyrosine) into 4-maleylacetoacetate, and homogentisic acid levels in the blood are a hundredfold higher than would normally be expected, despite the fact that a substantial amount is eliminated into the urine by the kidneys.[1]
The homogentisic acid is converted to the related substance benzoquinone acetic acid (BQA) which forms polymers that resemble the skin pigment melanin. These are deposited in the collagen, a connective tissue protein, of particular tissues such as cartilage. This process is called ochronosis (as the tissue looks ochre); ochronotic tissue is stiffened and unusually brittle, impairing its normal function and causing damage.[1]
Diagnosis

If the diagnosis of alkaptonuria is suspected, this can be confirmed or excluded by collecting urine for twenty-four hours and determining the amount of homogentisic acid by means of chromatography. There is no validated assay of HGA in blood.[1]
The severity of the symptoms and response to treatment can be quantified through a validated questionnaire titled the AKU Severity Score Index. This includes assigns scores to the presence of particular symptoms and features, such as the presence of eye and skin pigmentation, joint pain, heart problems and organ stones.[1]
Treatment
No treatment modality has been unequivocally demonstrated to reduce the complications of alkaptonuria. Main treatment attempts have focused on preventing ochronosis through the reduction of accumulating homogentisic acid. Such commonly recommended treatments include large doses of ascorbic acid (vitamin C) or dietary restriction of amino acids phenylalanine and tyrosine. However, vitamin C treatment has not shown to be effective,[1] and protein restriction (which can be difficult to adhere to) has not shown to be effective in clinical studies.[1]
Several recent studies have suggested that the herbicide nitisinone may be effective in the treatment of alkaptonuria. Nitisinone inhibits the enzyme, 4-hydroxyphenylpyruvate dioxygenase, responsible for converting tyrosine to homogentisic acid, thereby blocking the production and accumulation of HGA. Nitisinone has been used for some time at much higher doses in the treatment of type I tyrosinemia. Nitisinone treatment has been shown to cause a larger than 95% reduction in plasma and urinary HGA.[1] The main drawback is accumulation of tyrosine, the long-term risks of which are unknown; there is a particular concern about damage to the cornea of the eye. Long-term use would require frequent monitoring for complications.[1]
Prognosis
Alkaptonuria does not appear to affect life expectancy, although the last study on the topic is from 1985.[1] The main impact is on quality of life; many people with alkaptonuria have disabling symptoms such as pain, poor sleep and breathing symptoms. These generally start in the fourth decade. The average age at requiring joint replacement surgery is 50–55 years.[1]
Epidemiology
In most ethnic groups, the prevalence of alkaptonuria is between 1:100,000 and 1:250,000.[4] In Slovakia and the Dominican Republic the disease is much more common, with prevalence estimated at 1:19,000 people.[4] As for Slovakia, this is not the result of a single mutation but due to a group of 12 mutations in specific "hot spots" of the HGD gene.[4] The Slovakian clustering probably arose in a small area in the northwest of the country and spread after the 1950s due to migration.[4]
History
Alkaptonuria was one of the four diseases described by Sir Archibald Edward Garrod, as being the result of the accumulation of intermediates due to metabolic deficiencies. He linked ochronosis with the accumulation of alkaptans in 1902,[4][5] and his views on the subject, including its mode of heritance, were summarized in a 1908 Croonian Lecture at the Royal College of Physicians.[4][6][7]
The defect was narrowed down to homogentisic acid oxidase deficiency in a study published in 1958.[4][8] The genetic basis was elucidated in 1996, when HGD mutations were demonstrated.[4][9]
A 1977 study showed that an ochronotic Egyptian mummy had probably suffered from alkaptonuria.[10][11]
Research directions
Research collaborations by several national centres have been established to find a more definitive treatment for alkaptonuria. This has included studies on the use of nitisinone and investigations into antioxidants to inhibit ochronosis.[4] The ideal treatment would replace HGD enzyme function without accumulating other substances.[1]
See also
- Ochronosis
- List of cutaneous conditions
- List of radiographic findings associated with cutaneous conditions
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Ranganath LR, Jarvis JC, Gallagher JA (May 2013). "Recent advances in management of alkaptonuria (invited review; best practice article)". J. Clin. Pathol. 66 (5): 367–73. PMID 23486607. doi:10.1136/jclinpath-2012-200877.
- ↑ Speeckaert R, Van Gele M, Speeckaert MM, Lambert J, van Geel N (July 2014). "The biology of hyperpigmentation syndromes". Pigment Cell Melanoma Res. 27 (4): 512–24. PMID 24612852. doi:10.1111/pcmr.12235.
- ↑ Lindner, Moritz; Bertelmann, Thomas (2014-01-30). "On the ocular findings in ochronosis: a systematic review of literature". BMC Ophthalmology. 14 (1): 12. ISSN 1471-2415. PMC 3915032
 . PMID 24479547. doi:10.1186/1471-2415-14-12.
. PMID 24479547. doi:10.1186/1471-2415-14-12. - 1 2 3 4 5 6 7 8 9 10 11 12 13 Zatkova A (December 2011). "An update on molecular genetics of Alkaptonuria (AKU)". J. Inherit. Metab. Dis. 34 (6): 1127–36. PMID 21720873. doi:10.1007/s10545-011-9363-z.
- ↑ Garrod AE (1902). "The incidence of alkaptonuria: a study in clinical individuality". Lancet. 2 (4137): 1616–1620. doi:10.1016/S0140-6736(01)41972-6. Reproduced in Garrod AE (2002). "The incidence of alkaptonuria: a study in chemical individuality. 1902 classical article". Yale Journal of Biology and Medicine. 75 (4): 221–31. PMC 2588790 . PMID 12784973.
- ↑ Garrod AE (1908). "The Croonian lectures on inborn errors of metabolism: lecture II: alkaptonuria". Lancet. 2: 73–79. doi:10.1016/s0140-6736(01)78041-5.
- ↑ Garrod AE (1909). "Inborn errors of metabolism". Oxford University Press. OL 7116744M.
- ↑ La Du BN, Zannoni VG, Laster L, Seegmiller JE (1 January 1958). "The nature of the defect in tyrosine metabolism in alcaptonuria" (PDF). Journal of Biological Chemistry. 230 (1): 251–60. PMID 13502394.
- ↑ Fernández-Cañón JM, Granadino B, Beltrán-Valero de Bernabé D, et al. (1996). "The molecular basis of alkaptonuria". Nature Genetics. 14 (1): 19–24. PMID 8782815. doi:10.1038/ng0996-19.
- ↑ Stenn FF, Milgram JW, Lee SL, Weigand RJ, Veis A (1977). "Biochemical identification of homogentisic acid pigment in an ochronotic egyptian mummy". Science. 197 (4303): 566–8. PMID 327549. doi:10.1126/science.327549.
- ↑ Lee, SL.; Stenn, FF. (Jul 1978). "Characterization of mummy bone ochronotic pigment.". JAMA. 240 (2): 136–8. PMID 351220. doi:10.1001/jama.1978.03290020058024.