Baeyer–Villiger oxidation
| Baeyer-Villiger oxidation | |
|---|---|
| Named after | Adolf von Baeyer Victor Villiger |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | baeyer-villiger-oxidation |
| RSC ontology ID | RXNO:0000031 |
The Baeyer–Villiger oxidation is an organic reaction that forms an ester from a ketone or a lactone from a cyclic ketone, using peroxyacids or peroxides as the oxidant.[1] The reaction is named after Adolf von Baeyer and Victor Villiger who first reported the reaction in 1899.[1]

Reaction mechanism
In the first step of the reaction mechanism, the peroxyacid protonates the oxygen of the carbonyl group.[1] This makes the carbonyl group more susceptible to attack by the peroxyacid.[1] Next, the peroxyacid attacks the carbon of the carbonyl group forming what is known as the Criegee intermediate.[1] Through a concerted mechanism, one of the substituents on the ketone migrates to the oxygen of the peroxide group while a carboxylic acid leaves.[1] This migration step is thought to be the rate determining step.[2] Finally, deprotonation of the oxocarbenium ion produces the ester.[1]

The products of the Baeyer–Villiger oxidation are believed to be controlled through both primary and secondary stereoelectronic effects.[3] The primary stereoelectronic effect in the Baeyer–Villiger oxidation refers to the necessity of the oxygen-oxygen bond in the peroxide group to be antiperiplanar to the group that migrates.[3] This orientation facilitates optimum overlap of the 𝛔 orbital of the migrating group to the 𝛔* orbital of the peroxide group.[1] The secondary stereoelectronic effect refers to the necessity of the lone pair on the oxygen of the hydroxyl group to be antiperiplanar to the migrating group.[3] This allows for optimum overlap of the oxygen nonbonding orbital with the 𝛔* orbital of the migrating group.[4] This migration step is also (at least in silico) assisted by two or three peroxyacid units enabling the hydroxyl proton to shuttle to its new position.[5]
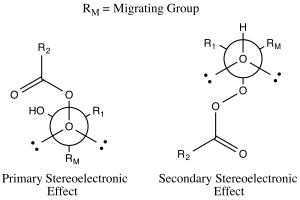
The migratory ability is ranked tertiary > secondary > aryl > primary.[6] Allylic groups are more apt to migrate than primary alkyl groups but less so than secondary alkyl groups.[4] Electron-withdrawing groups on the substituent decrease the rate of migration.[7] There are two explanations for this trend in migration ability.[8] One explanation relies on the buildup of positive charge in the transition state for breakdown of the Criegee intermediate (illustrated by the carbocation resonance structure of the Criegee intermediate).[8] Keeping this structure in mind, it makes sense that the substituent that can maintain positive charge the best would be most likely to migrate.[8] The higher the degree of substitution, the more stable a carbocation generally is.[9] Therefore, the tertiary > secondary > primary trend is observed.

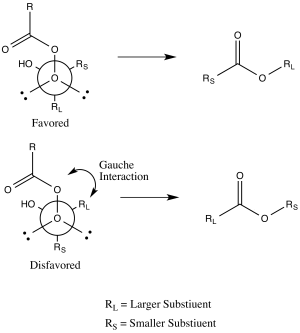
Another explanation uses stereoelectronic effects and steric arguments.[10] As mentioned, the substituent that is antiperiplanar to the peroxide group in the transition state will migrate.[3] This transition state has a gauche interaction between the peroxyacid and the non-migrating substituent.[10] If the bulkier group is placed antiperiplanar to the peroxide group, the gauche interaction between the substituent on the forming ester and the carbonyl group of the peroxyacid will be reduced.[10] Thus, it is the bulkier group that will prefer to be antiperiplanar to the peroxide group, enhancing its aptitude for migration.[10]
Historical background
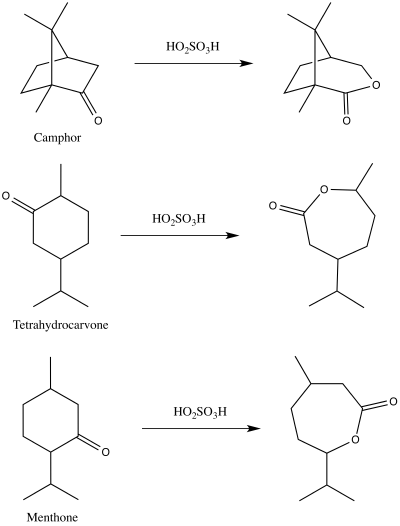
In 1899, Adolf Baeyer and Victor Villiger first published a demonstration of the reaction that we now know as the Baeyer–Villiger oxidation.[11][12] They used peroxymonosulfuric acid to make the corresponding lactones from camphor, menthone, and tetrahydrocarvone.[12][13]
There were three suggested reaction mechanisms of the Baeyer–Villiger oxidation that seemed to fit with observed reaction outcomes.[14] These three reaction mechanisms can really be split into two pathways of peroxyacid attack - on either the oxygen or the carbon of the carbonyl group.[15] Attack on oxygen could lead to two possible intermediates: Baeyer and Villiger suggested a dioxirane intermediate, while Georg Wittig and Gustav Pieper suggested a peroxide with no dioxirane formation.[15] Carbon attack was suggested by Rudolf Criegee.[15] In this pathway, the peracid attacks the carbonyl carbon, producing what is now known as the Criegee intermediate.[15]

In 1953, William von Eggers Doering and Edwin Dorfman elucidated the correct pathway for the reaction mechanism of the Baeyer–Villiger oxidation by using oxygen-18-labelling of benzophenone.[14] The three different mechanisms would each lead to a different distribution of labelled products. The Criegee intermediate would lead to a product only labelled on the carbonyl oxygen.[14] The product of the Wittig and Pieper intermediate is only labeled on the alkoxy group of the ester.[14] The Baeyer and Villiger intermediate leads to a 1:1 distribution of both of the above products.[14] The outcome of the labelling experiment supported the Criegee intermediate,[14] which is now the generally accepted pathway.[1]

Stereochemistry
The migration does not change the stereochemistry of the group that transfers, i.e.: it is stereoretentive.[16][17]
Reagents
Although many different peroxyacids are used for the Baeyer–Villiger oxidation, some of the more common oxidants include meta-chloroperbenzoic acid (mCPBA) and trifluoroperacetic acid (TFPAA).[2] The general trend is that higher reactivity is correlated with lower pKa (i.e.: stronger acidity) of the corresponding carboxylic acid (or alcohol in the case of the peroxides).[4] Therefore, the reactivity trend shows TFPAA > 4-nitroperbenzoic acid > mCPBA and performic acid > peracetic acid > hydrogen peroxide > tert-butyl hydroperoxide.[4] The peroxides are much less reactive than the peroxyacids.[2] The use of hydrogen peroxide even requires a catalyst.[6][18] In addition, using organic peroxides and hydrogen peroxide tends to generate more side-reactivity due to their promiscuity.[19]
Limitations
The use of peroxyacids and peroxides when performing the Baeyer–Villiger oxidation can cause the undesirable oxidation of other functional groups.[20] Alkenes and amines are a few of the groups that can be oxidized.[20] For instance, alkenes in the substrate, particularly when electron-rich, may be oxidized to epoxides.[20][21] However, methods have been developed that will allow for the tolerance of these functional groups.[20] In 1962, G. B. Payne reported that the use of hydrogen peroxide in the presence of a selenium catalyst will produce the epoxide from alkenyl ketones, while use of peroxyacetic acid will form the ester.[22]

Modifications
Catalytic Baeyer-Villiger oxidation
The use of hydrogen peroxide as an oxidant would be advantageous, making the reaction more environmentally friendly as the sole byproduct is water.[6] Benzeneseleninic acid derivatives as catalysts have been reported to give high selectivity with hydrogen peroxide as the oxidant.[23]
Baeyer-Villiger monooxygenases
Another way to create a catalytic Baeyer–Villiger oxidation is by using enzymes as the catalyst.[6] Baeyer-Villiger monooxygenases (BVMOs) use dioxygen to perform the Baeyer–Villiger oxidation.[6] These enzymes are capable of enantioselective oxidations of prochiral substrates.[6]
Asymmetric Baeyer-Villiger oxidation
There have been attempts to use organometallic catalysts to perform enantioselective Baeyer–Villiger oxidations.[6] The first reported instance of one such oxidation of a prochiral ketone used dioxygen as the oxidant with a copper catalyst.[21] Other catalysts, including platinum and aluminum compounds, followed.[21]
Applications
Zoapatanol
Zoapatanol is a biologically active molecule that occurs naturally in the zeopatle plant, which has been used in Mexico to make a tea that can induce menstruation and labor.[24] In 1981, Vinayak Kane and Donald Doyle reported a synthesis of zoapatanol.[25][26] They used the Baeyer–Villiger oxidation to make a lactone that served as a crucial building block that ultimately led to the synthesis of zoapatanol.[25][26]

Steroids
In 2013, Alina Świzdor reported the transformation of the steroid dehydroepiandrosterone to anticancer agent testololactone by use of a Baeyer-Villiger oxidation induced by fungus that produces Baeyer-Villiger monooxygenases.[27]

See also
References
- 1 2 3 4 5 6 7 8 9 Kürti, László; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Burlington; San Diego; London: Elsevier Academic Press. p. 28. ISBN 978-0-12-369483-6.
- 1 2 3 Krow, Grant R. (1993). "The Baeyer-Villiger Oxidation of Ketones and Aldehydes". Organic Reactions. 43 (3): 251–798. doi:10.1002/0471264180.or043.03.
- 1 2 3 4 Crudden, Cathleen M.; Chen, Austin C.; Calhoun, Larry A. (2000). "A Demonstration of the Primary Stereoelectronic Effect in the Baeyer-Villiger Oxidation of α-Fluorocyclohexanones". Angew. Chem. Int. Ed. 39 (16): 2851–2855. doi:10.1002/1521-3773(20000818)39:16<2851::aid-anie2851>3.0.co;2-y.
- 1 2 3 4 Myers, Andrew G. "Chemistry 115 Handouts: Oxidation" (PDF).
- ↑ The Role of Hydrogen Bonds in Baeyer-Villiger Reactions Shinichi Yamabe and Shoko Yamazaki J. Org. Chem.; 2007; 72(8) pp 3031–41; (Article) doi:10.1021/jo0626562
- 1 2 3 4 5 6 7 ten Brink, G.-J.; Arends, W. C. E.; Sheldon, R. A. (2004). "The Baeyer-Villiger Reaction: New Developments toward Greener Procedures". Chem. Rev. 104 (9): 4105–4123. doi:10.1021/cr030011l.
- ↑ Li, Jie Jack; Corey, E. J., eds. (2007). Name Reactions of Functional Group Transformations. Hoboken, NJ: Wiley-Interscience.
- 1 2 3 Hawthorne, M. Frederick; Emmons, William D.; McCallum, K. S. (1958). "A Re-examination of the Peroxyacid Cleavage of Ketones. I. Relative Migratory Aptitudes". J. Am. Chem. Soc. 80 (23): 6393–6398. doi:10.1021/ja01556a057.
- ↑ Jones, Jr., Maitland; Fleming, Steven A. (2010). Organic Chemistry (4th ed.). Canada: W. W. Norton & Company. p. 293. ISBN 978-0-393-93149-5.
- 1 2 3 4 Evans, D. A. "Stereoelectronic Effects-2" (PDF). Chemistry 206 (Fall 2006-2007).
- ↑ Baeyer, Adolf; Villiger, Victor (1899). "Einwirkung des Caro'schen Reagens auf Ketone". Ber. Dtsch. Chem. Ges. 32 (3): 3625–3633. doi:10.1002/cber.189903203151.
- 1 2 Hassall, C. H. (1957). "The Baeyer-Villiger Oxidation of Aldehydes and Ketones". Organic Reactions. 9 (3): 73–106. doi:10.1002/0471264180.or009.03.
- ↑ Renz, Michael; Meunier, Bernard (1999). "100 Years of Baeyer-Villiger Oxidations". Eur. J. Org. Chem. 1999 (4): 737–750. doi:10.1002/(SICI)1099-0690(199904)1999:4<737::AID-EJOC737>3.0.CO;2-B.
- 1 2 3 4 5 6 Doering, W. von E.; Dorfman, Edwin (1953). "Mechanism of the Peracid Ketone-Ester Conversion. Analysis of Organic Compounds for Oxygen-18". J. Am. Chem. Soc. 75 (22): 5595–5598. doi:10.1021/ja01118a035.
- 1 2 3 4 Doering, W. von E.; Speers, Louise (1950). "The Peracetic Acid Cleavage of Unsymmetrical Ketones". 72 (12): 5515–5518. doi:10.1021/ja01168a041.
- ↑ Turner, Richard B. (1950). "Stereochemistry of the Peracid Oxidation of Ketones". J. Am. Chem. Soc. 72 (2): 878–882. doi:10.1021/ja01158a061.
- ↑ Gallagher, T. F.; Kritchevsky, Theodore H. (1950). "Perbenzoic Acid Oxidation of 20-Ketosteroids and the Stereochemistry of C-17". J. Am. Chem. Soc. 72 (2): 882–885. doi:10.1021/ja01158a062.
- ↑ Cavarzan, Alessandra; Scarso, Alessandro; Sgarbossa, Paolo; Michelin, Rino A.; Strukul, Giorgio (2010). "Green Catalytic Baeyer–Villiger Oxidation with Hydrogen Peroxide in Water Mediated by Pt(II) Catalysts". ChemCatChem. 2 (10): 1296–1302. doi:10.1002/cctc.201000088.
- ↑ B. Schweitzer-Chaput, T. Kurtén, M. Klussmann, Angew. Chem. Int. Ed. 2015, 54, 11848–11851. doi:10.1002/anie.201505648
- 1 2 3 4 Grant R. Krow (1991). Trost, Barry M.; Fleming, Ian, eds. Comprehensive Organic Synthesis - Selectivity, Strategy and Efficiency in Modern Organic Chemistry, Volumes 1 - 9. Elsevier. pp. 671–688. ISBN 978-0-08-035930-4.
- 1 2 3 Seymour, Craig. "Page 1 The Asymmetric Baeyer-Villiger Oxidation" (PDF). http://www.scs.illinois.edu/denmark/wp-content/uploads/gp/2013/gm-2013-7-16.pdf. External link in
|website=(help) - ↑ Payne, G. B. (1962). "A Simplified Procedure for Epoxidation by Benzonitrile-Hydrogen Peroxide. Selective Oxidation of 2-Allylcyclohexanone". Tetrahedron. 18 (6): 763–765. doi:10.1016/S0040-4020(01)92726-7.
- ↑ ten Brink, Gerd-Jan; Vis, Jan-Martijn; Arends, Isabel W. C. E.; Sheldon, Roger A. (2001). "Selenium-Catalyzed Oxidations with Aqueous Hydrogen Peroxide. 2. Baeyer−Villiger Reactions in Homogeneous Solution". J. Org. Chem. 66 (7): 2429–2433. doi:10.1021/jo0057710.
- ↑ Levine, Seymour D.; Adams, Richard E.; Chen, Robert; Cotter, Mary Lou; Hirsch, Allen F.; Kane, Vinayak V.; Kanojia, Ramesh M.; Shaw, Charles; Wachter, Michael P.; Chin, Eva; Huettemann, Richard; Ostrowski, Paul (1979). "Zoapatanol and Montanol, Novel Oxepane Diterpenoids, from the Mexican Plant Zoapatle (Montanoa tomentosa)". J. Am. Chem. Soc. 101 (12): 3405–3407. doi:10.1021/ja00506a057.
- 1 2 Kane, Vinayak V.; Doyle, Donald L. (1981). "Total Synthesis of (±) Zoapatanol: A Stereospecific Synthesis of a Key Intermediate". Tetrahedron Lett. 22 (32): 3027–3030. doi:10.1016/S0040-4039(01)81818-9.
- 1 2 Kane, Vinayak V.; Doyle, Donald L. (1981). "Total Synthesis of (±) Zoapatanol". Tetrahedron Lett. 22 (32): 3031–3034. doi:10.1016/S0040-4039(01)81819-0.
- ↑ Świzdor, Alina (2013). "Baeyer-Villiger Oxidation of Some C19 Steroids by Penicillium lanosocoeruleum". Molecules. 18 (11): 13812–13822. doi:10.3390/molecules181113812.
{kind=link}