Aryl halide
In organic chemistry, an aryl halide (also known as haloarene or halogenoarene) is an aromatic compound in which one or more hydrogen atoms directly bonded to an aromatic ring are replaced by a halide. The haloarene are distinguished from haloalkanes because they exhibit many differences in methods of preparation and properties. The most important members are the aryl chlorides, but the class of compounds is so broad that many derivatives enjoy niche applications.
Preparation
The two main preparatory routes to aryl halides are direct halogenation and via diazonium salts.[1]
Direct halogenation
In the Friedel-Crafts halogenation, Lewis acids serve as catalysts. Many metal chlorides are used, examples include iron(III) chloride or aluminium chloride. The most important aryl halide, chlorobenzene is produced by this route. Monochlorination of benzene is always accompanied by formation of the dichlorobenzene derivatives.[2]
Arenes with electron donating groups react with halogens even in the absence of Lewis acids. For example, phenols and anilines react quickly with chlorine and bromine water to give multiple halogenated products.[3] The decolouration of bromine water by electron-rich arenes is used in the bromine test.
Direct halogenation of arenes are possible in the presence of light or at high temperature. For alkylbenzene derivatives, the alkyl positions tend to be halogenated first in the free radical halogenation. To halogenate the ring, Lewis acids are required, and light should be excluded to avoid the competing reaction.[1]

Sandmeyer, Schiemann and Gatterman reactions
The second main route is the Sandmeyer reaction. Anilines (aryl amines) are converted to their diazonium salts using nitrous acid. For example, copper(I) chloride converts diazonium salts to the aryl chloride. Nitrogen gas is the leaving group, which makes this reaction very favorable. The similar Schiemann reaction uses the tetrafluoroborate anion as the fluoride donor. Gatterman Reaction can also be used to convert Diazonium salt to chlorobenzene or bromobenzene by using copper powder instead of copper chloride or copper bromide. But this must be done in the presence of HCl and HBr respectively.
Halogenation in nature
Aryl halides occur widely in nature, most commonly produced by marine organisms that utilize the chloride and bromide in ocean waters. Chlorinated and brominated aromatic compounds are also numerous, e.g. derivatives of tyrosine, tryptophan, and various pyrrole derivatives. Some of these naturally occurring aryl halides exhibit useful medicinal properties.[4][5]

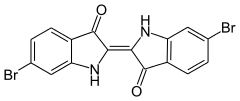
Structural trends
The C-X distances for aryl halides follow the expected trend. These distances for fluorobenzene, chlorobenzene, bromobenzene, and methyl 4-iodobenzoate are 135.6(4), 173.90(23), 189.8(1), and 209.9 pm, respectively.[6]
Reactions
Substitution
Unlike typical alkyl halides, aryl halides do not participate in conventional SN2 reactions, as the backside attack required for an SN2 reaction is impossible, owing to the planar structure of the aryl group. SN1 reactions are theoretically possible, but not generally observed, as the formation of the aryl cation is not energetically favourable.
However, aryl halides with electron-withdrawing groups in the ortho and para positions, can undergo SNAr reactions. For example, 2,4-dinitrochlorobenzene can react with water in basic solution to give a phenol:

Unlike in other substitution reactions, fluoride is the best leaving group, and iodide the worst, due to the fluoride's high electronegativity allowing better stabilization of the negatively charged intermediate.
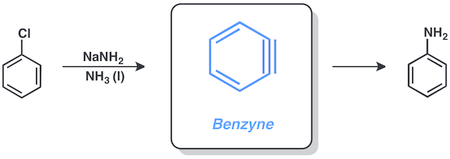
Benzyne
Aryl halides are capable of doing reactions via the benzyne mechanism, involving sodium amide in liquid ammonia. For example, chlorobenzene can be converted to aniline under these conditions.
Organometallic reagent formation
Aryl halides react with metals, generally lithium or magnesium, to give more reactive derivatives that behave as sources of aryl anions.
Direct formation of Grignard reagents, by adding the magnesium to the aryl halide in an ethereal solution, works well if the aromatic ring is not significantly deactivated by electron-withdrawing groups.
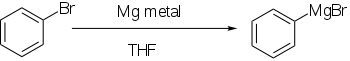
Compounds such as para-bromonitrobenzene cannot form stable Grignards directly, as their aromatic rings are too deactivated. If such a Grignard is needed, it's usually made by magnesium-halogen exchange involving isopropyl magnesium chloride at -78oC. This reaction takes place because the pKa of the aromatic protons is much lower—generally around 45, while that of the aliphatic alkane exceeds 50. The Grignards made using this procedure are generally used immediately, to avoid decomposition.

Other reactions
The halides can be displaced by strong nucleophiles via reactions involving radical anions. Alternatively aryl halides, especially the bromides and iodides, undergo oxidative addition, and thus are subject to Buchwald–Hartwig amination-type reactions.
Chlorobenzene was once the precursor to phenol, which is now made by oxidation of cumene. At high temperatures, aryl groups react with ammonia to give anilines.[2]
Biodegradation
Rhodococcus phenolicus is a bacterium species able to degrade dichlorobenzene as sole carbon sources.[7]
Applications
The aryl halides produced on the largest scale are chlorobenzene and the isomers of dichlorobenzene. One major but discontinued application was the use of chlorobenzene as a solvent for dispersing the herbicide Lasso. Overall, production of aryl chlorides as well as related naphthyl derivatives has been declining since the 1980s, in part due to environmental concerns.[2] Triphenylphosphine is produced from chlorobenzene:
- 3 C6H5Cl + PCl3 + 6 Na → P(C6H5)3 + 6 NaCl
Aryl bromides are widely used as fire-retardants. The most prominent member is tetrabromobisphenol-A, which is prepared by direct bromination of the diphenol.[8]
References
- 1 2 Boyd, Robert W.; Morrison, Robert (1992). Organic chemistry. Englewood Cliffs, N.J: Prentice Hall. p. 947. ISBN 0-13-643669-2.
- 1 2 3 Beck, U.; Löser, E. (2011). "Chlorinated Benzenes and Other Nucleus-Chlorinated Aromatic Hydrocarbons". Ullmann's Encyclopedia of Industrial Chemistry. ISBN 3527306730. doi:10.1002/14356007.o06_o03.
- ↑ Illustrative procedure for chlorination of an aromatic compound: Edward R. Atkinson; Donald M. Murphy; James E. Lufkin (1951). "dl-4,4',6,6'-Tetrachlorodiphenic Acid". Org. Synth.; Coll. Vol., 4, p. 872
- ↑ Fujimori, Danica Galonić; Walsh, Christopher T. (2007). "What's new in enzymatic halogenations". Current Opinion in Chemical Biology. 11 (5): 553–60. PMC 2151916
 . PMID 17881282. doi:10.1016/j.cbpa.2007.08.002.
. PMID 17881282. doi:10.1016/j.cbpa.2007.08.002. - ↑ Gribble, Gordon W. (2004). "Natural Organohalogens: A New Frontier for Medicinal Agents?". Journal of Chemical Education. 81 (10): 1441. Bibcode:2004JChEd..81.1441G. doi:10.1021/ed081p1441.
- ↑ Oberhammer, Heinz (2009). "The Structural Chemistry of Carbon-Halogen Bonds". PATai's Chemistry of Functional Groups. ISBN 978-0-470-68253-1. doi:10.1002/9780470682531.pat0002.
- ↑ Rehfuss, Marc; Urban, James (2005). "Rhodococcus phenolicus sp. nov., a novel bioprocessor isolated actinomycete with the ability to degrade chlorobenzene, dichlorobenzene and phenol as sole carbon sources". Systematic and Applied Microbiology. 28 (8): 695–701. PMID 16261859. doi:10.1016/j.syapm.2005.05.011.
- ↑ Ioffe, D.; Kampf, A. (2002). "Bromine, Organic Compounds". Kirk-Othmer Encyclopedia of Chemical Technology. ISBN 0471238961. doi:10.1002/0471238961.0218151325150606.a01..