Acid–base reaction
| Acids and bases |
|---|
| Acid types |
| Base types |
An acid–base reaction is a chemical reaction that occurs between an acid and a base. Several theoretical frameworks provide alternative conceptions of the reaction mechanisms and their application in solving related problems; these are called acid–base theories, for example, Brønsted–Lowry acid–base theory. Their importance becomes apparent in analyzing acid–base reactions for gaseous or liquid species, or when acid or base character may be somewhat less apparent. The first of these concepts was provided by the French chemist Antoine Lavoisier, around 1776.[1]
Acid–base definitions
Historic development
Lavoisier's oxygen theory of acids
The first scientific concept of acids and bases was provided by Lavoisier in around 1776. Since Lavoisier's knowledge of strong acids was mainly restricted to oxoacids, such as HNO
3 (nitric acid) and H
2SO
4 (sulfuric acid), which tend to contain central atoms in high oxidation states surrounded by oxygen, and since he was not aware of the true composition of the hydrohalic acids (HF, HCl, HBr, and HI), he defined acids in terms of their containing oxygen, which in fact he named from Greek words meaning "acid-former" (from the Greek οξυς (oxys) meaning "acid" or "sharp" and γεινομαι (geinomai) meaning "engender"). The Lavoisier definition was held as absolute truth for over 30 years, until the 1810 article and subsequent lectures by Sir Humphry Davy in which he proved the lack of oxygen in H
2S, H2Te, and the hydrohalic acids. However, Davy failed to develop a new theory, concluding that "acidity does not depend upon any particular elementary substance, but upon peculiar arrangement of various substances".[2] One notable modification of oxygen theory was provided by Berzelius, who stated that acids are oxides of nonmetals while bases are oxides of metals.
Liebig's hydrogen theory of acids
In 1838, Justus von Liebig proposed that an acid is a hydrogen-containing substance in which the hydrogen could be replaced by a metal.[3][4][5] This redefinition was based on his extensive work on the chemical composition of organic acids, finishing the doctrinal shift from oxygen-based acids to hydrogen-based acids started by Davy. Liebig's definition, while completely empirical, remained in use for almost 50 years until the adoption of the Arrhenius definition.[6]
Arrhenius definition

The first modern definition of acids and bases in molecular terms was devised by Svante Arrhenius.[7][8] A hydrogen theory of acids, it followed from his 1884 work with Friedrich Wilhelm Ostwald in establishing the presence of ions in aqueous solution and led to Arrhenius receiving the Nobel Prize in Chemistry in 1903.
As defined by Arrhenius:
- an Arrhenius acid is a substance that dissociates in water to form hydrogen ions (H+);[9] that is, an acid increases the concentration of H+ ions in an aqueous solution.
This causes the protonation of water, or the creation of the hydronium (H3O+) ion.[note 1] Thus, in modern times, the symbol H+ is interpreted as a shorthand for H3O+, because it is now known that a bare proton does not exist as a free species in aqueous solution.[12]
- an Arrhenius base is a substance that dissociates in water to form hydroxide (OH−) ions; that is, a base increases the concentration of OH− ions in an aqueous solution.
The Arrhenius definitions of acidity and alkalinity are restricted to aqueous solutions, and refer to the concentration of the solvent ions. Under this definition, pure H2SO4 and HCl dissolved in toluene are not acidic, and molten NaOH and solutions of calcium amide in liquid ammonia are not alkaline.
Overall, to qualify as an Arrhenius acid, upon the introduction to water, the chemical must either cause, directly or otherwise:
- an increase in the aqueous hydronium concentration, or
- a decrease in the aqueous hydroxide concentration.
Conversely, to qualify as an Arrhenius base, upon the introduction to water, the chemical must either cause, directly or otherwise:
- a decrease in the aqueous hydronium concentration, or
- an increase in the aqueous hydroxide concentration.
The reaction of an acid with a base is called a neutralization reaction. The products of this reaction are a salt and water.
- acid + base → salt + water
In this traditional representation an acid–base neutralization reaction is formulated as a double-replacement reaction. For example, the reaction of hydrochloric acid, HCl, with sodium hydroxide, NaOH, solutions produces a solution of sodium chloride, NaCl, and some additional water molecules.
- HCl(aq) + NaOH(aq) → NaCl(aq) + H2O
The modifier (aq) in this equation is important. It was implied by Arrhenius, not included explicitly. It indicates that the substances are dissolved in water. In fact though all three substances, HCl, NaOH and NaCl are capable of existing as pure compounds, in aqueous solutions they are fully dissociated into the (aquated) ions H+, Cl−, Na+ and OH−.
Brønsted–Lowry definition
  | |
| Johannes Nicolaus Brønsted and Thomas Martin Lowry |
The Brønsted–Lowry definition, formulated in 1923, independently by Johannes Nicolaus Brønsted in Denmark and Martin Lowry in England,[13][14] is based upon the idea of protonation of bases through the de-protonation of acids – that is, the ability of acids to "donate" hydrogen ions (H+)—otherwise known as protons—to bases, which "accept" them.[15][note 2]
An acid–base reaction is, thus, the removal of a hydrogen ion from the acid and its addition to the base.[16] The removal of a hydrogen ion from an acid produces its conjugate base, which is the acid with a hydrogen ion removed. The reception of a proton by a base produces its conjugate acid, which is the base with a hydrogen ion added.
Unlike the previous definitions, the Brønsted–Lowry definition does not refer to the formation of salt and solvent, but instead to the formation of conjugate acids and conjugate bases, produced by the transfer of a proton from the acid to the base.[9][15] In this approach, acids and bases are fundamentally different in behavior from salts, which are seen as electrolytes, subject to the theories of Debye, Onsager, and others. An acid and a base react not to produce a salt and a solvent, but to form a new acid and a new base. The concept of neutralization is thus absent.[2] Brønsted–Lowry acid–base behavior is formally independent of any solvent, making it more all-encompassing than the Arrhenius model.
The general formula for acid–base reactions according to the Brønsted–Lowry definition is:
- HA + B → BH+ + A−
where HA represents the acid, B represents the base, BH+ represents the conjugate acid of B, and A− represents the conjugate base of HA.
For example, a Brønsted-Lowry model for the dissociation of hydrochloric acid (HCl) in aqueous solution would be the following:
- HCl + H2O ⇌ H3O+ + Cl−
The removal of H+ from the HCl produces the chloride ion, Cl−, the conjugate base of the acid. The addition of H+ to the H2O (acting as a base) forms the hydronium ion, H3O+, the conjugate acid of the base.
Water is amphoteric—that is, it can act as both an acid and a base. The Brønsted-Lowry model explains this, showing the dissociation of water into low concentrations of hydronium and hydroxide ions:
- H2O + H2O ⇌ H3O+ + OH−
This equation is demonstrated in the image below:
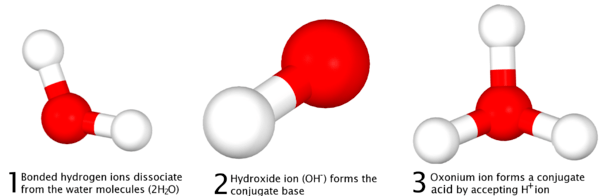
Here, one molecule of water acts as an acid, donating an H+ and forming the conjugate base, OH−, and a second molecule of water acts as a base, accepting the H+ ion and forming the conjugate acid, H3O+.
As an example of water acting as an acid, consider an aqueous solution of pyridine, C5H5N.
- C5H5N + H2O ⇌ [C5H5NH]+ + OH−
In this example, a water molecule is split into a hydrogen ion, which is donated to a pyridine molecule, and an hydroxide ion.
In the Brønsted-Lowry model, the solvent does not necessarily have to be water. For example, consider what happens when acetic acid, CH3COOH, dissolves in liquid ammonia.
- CH
3COOH + NH
3 ⇌ NH+
4 + CH
3COO−
An H+ ion is removed from acetic acid, forming its conjugate base, the acetate ion, CH3COO−. The addition of an H+ ion to an ammonia molecule of the solvent creates its conjugate acid, the ammonium ion, NH+
4.
The Brønsted–Lowry model calls hydrogen-containing substances (like HCl) acids. Thus, some substances, which many chemists considered to be acids, such as SO3 or BCl3, are excluded from this classification due to lack of hydrogen. Gilbert N. Lewis wrote in 1938, "To restrict the group of acids to those substances that contain hydrogen interferes as seriously with the systematic understanding of chemistry as would the restriction of the term oxidizing agent to substances containing oxygen."[2] Furthermore, KOH and KNH2 are not considered Brønsted bases, but rather salts containing the bases OH− and NH−
2.
Lewis definition
The hydrogen requirement of Arrhenius and Brønsted–Lowry was removed by the Lewis definition of acid–base reactions, devised by Gilbert N. Lewis in 1923,[17] in the same year as Brønsted–Lowry, but it was not elaborated by him until 1938.[2] Instead of defining acid–base reactions in terms of protons or other bonded substances, the Lewis definition defines a base (referred to as a Lewis base) to be a compound that can donate an electron pair, and an acid (a Lewis acid) to be a compound that can receive this electron pair.[18]
For example, boron trifluoride, BF3 is a typical Lewis acid. It can accept a pair of electrons as it has a vacancy in its octet. The fluoride ion has a full octet and can donate a pair of electrons. Thus
- BF3 + F− → BF−
4
is a typical Lewis acid, Lewis base reaction. All compounds of group 13 elements with a formula AX3 can behave as Lewis acids. Similarly, compounds of group 15 elements with a formula DY3, such as amines, NR3, and phosphines, PR3, can behave as Lewis bases. Adducts between them have the formula X3A←DY3 with a dative covalent bond, shown symbolically as ←, between the atoms A (acceptor) and D (donor). Compounds of group 16 with a formula DX2 may also act as Lewis bases; in this way, a compound like an ether, R2O, or a thioether, R2S, can act as a Lewis base. The Lewis definition is not limited to these examples. For instance, carbon monoxide acts as a Lewis base when it forms an adduct with boron trifluoride, of formula F3B←CO
Adducts involving metal ions are referred to as co-ordination compounds; each ligand donates a pair of electrons to the metal ion.[18] The reaction
- [Ag(H2O)4]+ + 2NH3 → [Ag(NH3)2]+ + 4H2O
can be seen as an acid–base reaction in which a stronger base (ammonia) replaces a weaker one (water)
The Lewis and Brønsted–Lowry definitions are consistent with each other since the reaction
- H+ + OH− ⇌ H2O
is an acid–base reaction in both theories.
Solvent system definition
One of the limitations of the Arrhenius definition is its reliance on water solutions. Edward Curtis Franklin studied the acid–base reactions in liquid ammonia in 1905 and pointed out the similarities to the water-based Arrhenius theory. Albert F. O. Germann, working with liquid phosgene, COCl
2, formulated the solvent-based theory in 1925, thereby generalizing the Arrhenius definition to cover aprotic solvents.[19]
Germann pointed out that in many solutions, there are ions in equilibrium with the neutral solvent molecules:
- solvonium:[note 3] A generic name for a positive ion.
- solvate:[note 4] A generic name for a negative ion.
For example, water and ammonia undergo such dissociation into hydronium and hydroxide, and ammonium and amide, respectively:
- 2 H
2O ⇌ H
3O+
+ OH− - 2 NH
3 ⇌ NH+
4 + NH−
2
Some aprotic systems also undergo such dissociation, such as dinitrogen tetroxide into nitrosonium and nitrate, antimony trichloride into dichloroantimonium and tetrachloroantimonate, and phosgene into chlorocarboxonium and chloride:
- N
2O
4 ⇌ NO+
+ NO−
3 - 2 SbCl
3 ⇌ SbCl+
2 + SbCl−
4 - COCl
2 ⇌ COCl+
+ Cl−
A solute that causes an increase in the concentration of the solvonium ions and a decrease in the concentration of solvate ions is defined as an acid. A solute that causes an increase in the concentration of the solvate ions and a decrease in the concentration of the solvonium ions is defined as a base.
Thus, in liquid ammonia, KNH
2 (supplying NH−
2) is a strong base, and NH
4NO
3 (supplying NH+
4) is a strong acid. In liquid sulfur dioxide (SO
2), thionyl compounds (supplying SO2+
) behave as acids, and sulfites (supplying SO2−
3) behave as bases.
The non-aqueous acid–base reactions in liquid ammonia are similar to the reactions in water:
- + → Na
2[Zn(NH
2)
4] - + → [Zn(NH
2)
4)]I
2
Nitric acid can be a base in liquid sulfuric acid:
- + 2 H
2SO
4 → NO+
2 + H
3O+
+ 2 HSO−
4
The unique strength of this definition shows in describing the reactions in aprotic solvents; for example, in liquid N
2O
4:
- + → +
Because the solvent system definition depends on the solute as well as on the solvent itself, a particular solute can be either an acid or a base depending on the choice of the solvent: HClO
4 is a strong acid in water, a weak acid in acetic acid, and a weak base in fluorosulfonic acid; this characteristic of the theory has been seen as both a strength and a weakness, because some substances (such as SO
3 and NH
3) have been seen to be acidic or basic on their own right. On the other hand, solvent system theory has been criticized as being too general to be useful. Also, it has been thought that there is something intrinsically acidic about hydrogen compounds, a property not shared by non-hydrogenic solvonium salts.[2]
Lux–Flood definition
This acid–base theory was a revival of oxygen theory of acids and bases, proposed by German chemist Hermann Lux[20][21] in 1939, further improved by Håkon Flood circa 1947[22] and is still used in modern geochemistry and electrochemistry of molten salts. This definition describes an acid as an oxide ion (O2−
) acceptor and a base as an oxide ion donor. For example:[23]
- + → MgCO
3 - + → CaSiO
3 - + → NO+
2 + 2 SO2−
4
This theory is also useful in the systematisation of the reactions of noble gas compounds, especially the xenon oxides, fluorides, and oxofluorides.[24]
Usanovich definition
Mikhail Usanovich developed a general theory that does not restrict acidity to hydrogen-containing compounds, but his approach, published in 1938, was even more general than Lewis theory.[2] Usanovich's theory can be summarized as defining an acid as anything that accepts negative species or donates positive ones, and a base as the reverse. This defined the concept of redox (oxidation-reduction) as a special case of acid–base reactions
Some examples of Usanovich acid–base reactions include:
- + → 2 Na+
+ SO2−
4 (species exchanged: O2−
anion) - + → 6 NH+
4 + 2 SbS3−
4 (species exchanged: 3 S2−
anions) - + → 2Na+
+ 2Cl−
(species exchanged: 2 electrons)
Pearson definition
In 1963, Ralph Pearson proposed a qualitative concept known as Hard Soft Acid Base principle.[25] later made quantitative with help of Robert Parr in 1984.[26][27] 'Hard' applies to species that are small, have high charge states, and are weakly polarizable. 'Soft' applies to species that are large, have low charge states and are strongly polarizable. Acids and bases interact, and the most stable interactions are hard–hard and soft–soft. This theory has found use in organic and inorganic chemistry.
Acid–base equilibrium
The reaction of a strong acid with a strong base is essentially a quantitative reaction. For example,
- HCl(aq) + Na(OH)(aq) → H2O + NaCl(aq)
In this reaction both the sodium and chloride ions are spectators as the neutralization reaction,
- H+ + OH− → H2O
does not involve them. With weak bases addition of acid is not quantitative because a solution of a weak base is a buffer solution. A solution of a weak acid is also a buffer solution. When a weak acid reacts with a weak base an equilibrium mixture is produced. For example, adenine, written as AH can react with a hydrogen phosphate ion, HPO2−
4.
- AH + HPO2−
4 ⇌ A− + H
2PO−
4
The equilibrium constant for this reaction can be derived from the acid dissociation constants of adenine and of the dihydrogen phosphate ion.
- [A−][H+] = Ka1[AH]
- [HPO2−
4][H+] = Ka2[H
2PO−
4]
The notation [X] signifies "concentration of X". When these two equations are combined by eliminating the hydrogen ion concentration, an expression for the equilibrium constant, K is obtained.
- [A−][H
2PO−
4] = K[AH][HPO2−
4]; K = Ka1/Ka2
Acid–alkali reaction
An acid–alkali reaction is a special case of an acid–base reaction, where the base used is also an alkali. When an acid reacts with an alkali salt (a metal hydroxide), the product is a metal salt and water. Acid–alkali reactions are also neutralization reactions.
In general, acid–alkali reactions can be simplified to
by omitting spectator ions.
Acids are in general pure substances that contain hydrogen cations (H+
) or cause them to be produced in solutions. Hydrochloric acid (HCl) and sulfuric acid (H
2SO
4) are common examples. In water, these break apart into ions:
- HCl → H+
(aq) + Cl−
(aq) - H
2SO
4 → H+
(aq) + HSO−
4(aq)
The alkali breaks apart in water, yielding dissolved hydroxide ions:
- NaOH → Na+
(aq) + OH−
(aq)
See also
- Acid–base titration
- Electron configuration
- Lewis structure
- Nucleophilic substitution and Redox reactions
- Protonation and Deprotonation
- Resonance structure
Notes
- ↑ More recent IUPAC recommendations now suggest the newer term "hydronium"[10] be used in favor of the older accepted term "oxonium"[11] to illustrate reaction mechanisms such as those defined in the Brønsted–Lowry and solvent system definitions more clearly, with the Arrhenius definition serving as a simple general outline of acid–base character.[9]
- ↑ "Removal of a proton from the nucleus of an atom does not occur - it would require very much more energy than is involved in the dissociation of acids."
- ↑ The term solvonium has replaced the older term lyonium.
- ↑ the term solvate has replaced the older term lyate.
References
- ↑ Miessler & Tarr 1991, p. 166 – Table of discoveries attributes Antoine Lavoisier as the first to posit a scientific theory in relation to oxyacids.
- 1 2 3 4 5 6 Hall, Norris F. (March 1940). "Systems of Acids and Bases". Journal of Chemical Education. 17 (3): 124–128. Bibcode:1940JChEd..17..124H. doi:10.1021/ed017p124.
- ↑ Miessler & Tarr 1991
- ↑ Meyers 2003, p. 156
- ↑ Miessler & Tarr 1991, p. 166 – table of discoveries attributes Justus von Liebig's publication as 1838
- ↑ Finston & Rychtman 1983, pp. 140–146
- ↑ Miessler G.L. and Tarr D.A. Inorganic Chemistry (2nd ed., Prentice-Hall 1999) p.154 ISBN 0-13-841891-8
- ↑ Whitten K.W., Galley K.D. and Davis R.E. General Chemistry (4th ed., Saunders 1992) p.356 ISBN 0-03-072373-6
- 1 2 3 Miessler & Tarr 1991, p. 165
- ↑ Murray, Kermit K.; Boyd, Robert K.; Eberlin, Marcos N.; Langley, G. John; Li, Liang; Naito, Yasuhide (June 2013) [2006]. "Standard definition of terms relating to mass spectrometry recommendations". Pure and Applied Chemistry. International Union of Pure and Applied Chemistry. 85 (7). doi:10.1351/PAC-REC-06-04-06. (In this document, there is no reference to deprecation of "oxonium", which is also still accepted, as it remains in the IUPAC Gold book, but rather reveals preference for the term "Hydronium".)
- ↑ "oxonium ylides". IUPAC Compendium of Chemical Terminology (interactive version) (2.3.3 ed.). International Union of Pure and Applied Chemistry. 2014-02-24. Retrieved 9 May 2007.
- ↑ LeMay, Eugene (2002). Chemistry. Upper Saddle River, New Jersey: Prentice-Hall. p. 602. ISBN 0-13-054383-7.
- ↑ Brönsted, J. N. (1923). "Einige Bemerkungen über den Begriff der Säuren und Basen" [Some observations about the concept of acids and bases]. Recueil des Travaux Chimiques des Pays-Bas. 42 (8): 718–728.
- ↑ Lowry, T. M. (1923). "The uniqueness of hydrogen". Journal of the Society of Chemical Industry. 42 (3): 43–47.
- 1 2 Miessler & Tarr 1991, pp. 167–169 – According to this page, the original definition was that "acids have a tendency to lose a proton"
- ↑ Clayden et al. 2000, pp. 182–184
- ↑ Miessler & Tarr 1991, p. 166 – Table of discoveries attributes the date of publication/release for the Lewis theory as 1923.
- 1 2 Miessler & Tarr 1991, pp. 170–172
- ↑ Germann, Albert F. O. (6 October 1925). "A General Theory of Solvent Systems". Journal of the American Chemical Society. 47 (10): 2461–2468. doi:10.1021/ja01687a006.
- ↑ Franz, H. (1966). "Solubility of Water Vapor in Alkali Borate Melts". Journal of the American Ceramic Society. 49 (9): 473–477. doi:10.1111/j.1151-2916.1966.tb13302.x.
- ↑ Lux, Hermann (1939). ""Säuren" und "Basen" im Schmelzfluss: die Bestimmung. der Sauerstoffionen-Konzentration". Z. Elektrochem. (in German). 45 (4): 303–309.
- ↑ Flood, H.; Forland, T. (1947). "The Acidic and Basic Properties of Oxides". Acta Chemica Scandinavica. 1 (6): 592–604. PMID 18907702. doi:10.3891/acta.chem.scand.01-0592.
- ↑ Drago, Russel S.; Whitten, Kenneth W. (1966). "The Synthesis of Oxyhalides Utilizing Fused-Salt Media". Inorganic Chemistry. 5 (4): 677–682. doi:10.1021/ic50038a038.
- ↑ Greenwood, Norman N.; Earnshaw, Alan (1984). Chemistry of the Elements. Oxford: Pergamon Press. p. 1056. ISBN 0-08-022057-6.
- ↑ Pearson, Ralph G. (1963). "Hard and Soft Acids and Bases.". Journal of the American Chemical Society. 85 (22): 3533–3539. doi:10.1021/ja00905a001.
- ↑ Parr, Robert G.; Pearson, Ralph G. (1983). "Absolute hardness: companion parameter to absolute electronegativity". Journal of the American Chemical Society. 105 (26): 7512–7516. doi:10.1021/ja00364a005.
- ↑ Pearson, Ralph G. (2005). "Chemical hardness and density functional theory" (PDF). Journal of Chemical Sciences. 117 (5): 369–377. doi:10.1007/BF02708340.
Sources
- Clayden, Jonathan; Greeves, Nick; Warren, Stuart; Wothers, Peter (2000). Organic Chemistry (First ed.). Oxford University Press.
- Finston, H. L.; Rychtman, A. C. (1983). A New View of Current Acid-Base Theories. New York: John Wiley & Sons.
- Meyers, R. (2003). The Basics of Chemistry. Greenwood Press.
- Miessler, G. L.; Tarr, D. A. (1991). Inorganic Chemistry.
External links
- Acid-base Physiology: an on-line text
- John W. Kimball's online Biology book section of acid and bases.