Alkali metal
| Alkali metals | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ↓ Period | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 |  3 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | .jpg) 11 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 |  19 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 |  37 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 |  55 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | Francium (Fr) 87 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Legend
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The alkali metals are a group (column) in the periodic table consisting of the chemical elements lithium (Li), sodium (Na), potassium (K),[note 1] rubidium (Rb), caesium (Cs),[note 2] and francium (Fr). This group lies in the s-block of the periodic table of elements as all alkali metals have their outermost electron in an s-orbital: this shared electron configuration results in them having very similar characteristic properties. Indeed, the alkali metals provide the best example of group trends in properties in the periodic table, with elements exhibiting well-characterised homologous behaviour.
The alkali metals are all shiny, soft, highly reactive metals at standard temperature and pressure and readily lose their outermost electron to form cations with charge +1. They can all be cut easily with a knife due to their softness, exposing a shiny surface that tarnishes rapidly in air due to oxidation by atmospheric moisture and oxygen (and in the case of lithium, nitrogen). Because of their high reactivity, they must be stored under oil to prevent reaction with air, and are found naturally only in salts and never as the free elements. Caesium, the fifth alkali metal, is the most reactive of all the metals. In the modern IUPAC nomenclature, the alkali metals comprise the group 1 elements,[note 3] excluding hydrogen (H), which is nominally a group 1 element but not normally considered to be an alkali metal as it rarely exhibits behaviour comparable to that of the alkali metals. All the alkali metals react with water, with the heavier alkali metals reacting more vigorously than the lighter ones.
All of the discovered alkali metals occur in nature as their compounds: in order of abundance, sodium is the most abundant, followed by potassium, lithium, rubidium, caesium, and finally francium, which is very rare due to its extremely high radioactivity; francium occurs only in the minutest traces in nature as an intermediate step in some obscure side branches of the natural decay chains. Experiments have been conducted to attempt the synthesis of ununennium (Uue), which is likely to be the next member of the group, but they have all met with failure. However, ununennium may not be an alkali metal due to relativistic effects, which are predicted to have a large influence on the chemical properties of superheavy elements; even if it does turn out to be an alkali metal, it is predicted to have some differences in physical and chemical properties from its lighter homologues.
Most alkali metals have many different applications. One of the best-known applications of the pure elements is the use of rubidium and caesium in atomic clocks, of which caesium atomic clocks are the most accurate and precise representation of time. A common application of the compounds of sodium is the sodium-vapour lamp, which emits very efficient light. Table salt, or sodium chloride, has been used since antiquity. Sodium and potassium are also essential elements, having major biological roles as electrolytes, and although the other alkali metals are not essential, they also have various effects on the body, both beneficial and harmful.
Properties
Physical and chemical
The physical and chemical properties of the alkali metals can be readily explained by their having an ns1 valence electron configuration, which results in weak metallic bonding. Hence, all the alkali metals are soft and have low densities,[5] melting[5] and boiling points,[5] as well as heats of sublimation, vaporisation, and dissociation.[6]:74 They all crystallise in the body-centered cubic crystal structure,[6]:73 and have distinctive flame colours because their outer s electron is very easily excited.[6]:75 The ns1 configuration also results in the alkali metals having very large atomic and ionic radii, as well as very high thermal and electrical conductivity.[6]:75 Their chemistry is dominated by the loss of their lone valence electron in the outermost s-orbital to form the +1 oxidation state, due to the ease of ionising this electron and the very high second ionisation energy.[6]:76 Most of the chemistry has been observed only for the first five members of the group. The chemistry of francium is not well established due to its extreme radioactivity;[5] thus, the presentation of its properties here is limited. What little is known about francium shows that it is very close in behaviour to caesium, as expected. The physical properties of francium are even sketchier because the bulk element has never been observed; hence any data that may be found in the literature are certainly speculative extrapolations.[7]
| Name | Lithium | Sodium | Potassium | Rubidium | Caesium | Francium |
|---|---|---|---|---|---|---|
| Atomic number | 3 | 11 | 19 | 37 | 55 | 87 |
| Standard atomic weight (u)[note 4][10][11] | 6.94(1)[note 5] | 22.98976928(2) | 39.0983(1) | 85.4678(3) | 132.9054519(2) | [223][note 6] |
| Electron configuration | [He] 2s1 | [Ne] 3s1 | [Ar] 4s1 | [Kr] 5s1 | [Xe] 6s1 | [Rn] 7s1 |
| Melting point (°C) | 180.54 | 97.72 | 63.38 | 39.31 | 28.44 | ? |
| Boiling point (°C) | 1342 | 883 | 759 | 688 | 671 | ? |
| Density (g·cm−3) | 0.534 | 0.968 | 0.89 | 1.532 | 1.93 | ? |
| Heat of fusion (kJ·mol−1) | 3.00 | 2.60 | 2.321 | 2.19 | 2.09 | ? |
| Heat of vaporisation (kJ·mol−1) | 136 | 97.42 | 79.1 | 69 | 66.1 | ? |
| Heat of formation of monatomic gas (kJ·mol−1) | 162 | 108 | 89.6 | 82.0 | 78.2 | ? |
| Electrical resistivity at 25 °C (nΩ·cm) | 94.7 | 48.8 | 73.9 | 131 | 208 | ? |
| Atomic radius (pm) | 152 | 186 | 227 | 248 | 265 | ? |
| Ionic radius of hexacoordinate M+ ion (pm) | 76 | 102 | 138 | 152 | 167 | ? |
| First ionisation energy (kJ·mol−1) | 520.2 | 495.8 | 418.8 | 403.0 | 375.7 | 392.8[12] |
| Electron affinity (kJ·mol−1) | 59.62 | 52.87 | 48.38 | 46.89 | 45.51 | ? |
| Enthalpy of dissociation of M2 (kJ·mol−1) | 106.5 | 73.6 | 57.3 | 45.6 | 44.77 | ? |
| Pauling electronegativity | 0.98 | 0.93 | 0.82 | 0.82 | 0.79 | ?[note 7] |
| Standard electrode potential (E°(M+→M0); V)[15] | −3.04 | −2.71 | −2.93 | −2.98 | −3.03 | ? |
| Flame test colour Principal emission/absorption wavelength (nm) |
Crimson 670.8 | Yellow 589.2 | Violet 766.5 | Red-violet 780.0 | Blue 455.5 | ? |
The alkali metals are more similar to each other than the elements in any other group are to each other.[5] Indeed, the similarity is so great that it is quite difficult to separate potassium, rubidium, and caesium, due to their similar ionic radii; lithium and sodium are more distinct. For instance, when moving down the table, all known alkali metals show increasing atomic radius,[16] decreasing electronegativity,[16] increasing reactivity,[5] and decreasing melting and boiling points[16] as well as heats of fusion and vaporisation.[6]:75 In general, their densities increase when moving down the table, with the exception that potassium is less dense than sodium.[16] One of the very few properties of the alkali metals that does not display a very smooth trend is their reduction potentials: lithium's value is anomalous, being more negative than the others.[6]:75 This is because the Li+ ion has a very high hydration energy in the gas phase: though the lithium ion disrupts the structure of water significantly, causing a higher change in entropy, this high hydration energy is enough to make the reduction potentials indicate it as being the most electropositive alkali metal, despite the difficulty of ionising it in the gas phase.[6]:75
The stable alkali metals are all silver-coloured metals except for caesium, which has a pale golden tint:[17] it is one of only three metals that are clearly coloured (the other two being copper and gold).[6]:74 Additionally, the heavy alkaline earth metals calcium, strontium, and barium, as well as the divalent lanthanides europium and ytterbium, are pale yellow, though the colour is much less prominent than it is for caesium.[6]:74 Their lustre tarnishes rapidly in air due to oxidation.[5] They all crystallise in the body-centered cubic crystal structure,[6]:73 and have distinctive flame colours because their outer s electron is very easily excited. Indeed, these flame test colours are the most common way of identifying them since all their salts with common ions are soluble.[6]:75
All the alkali metals are highly reactive and are never found in elemental forms in nature.[18] Because of this, they are usually stored in mineral oil or kerosene (paraffin oil).[19] They react aggressively with the halogens to form the alkali metal halides, which are white ionic crystalline compounds that are all soluble in water except lithium fluoride (LiF).[5] The alkali metals also react with water to form strongly alkaline hydroxides and thus should be handled with great care. The heavier alkali metals react more vigorously than the lighter ones; for example, when dropped into water, caesium produces a larger explosion than potassium if the same number of moles of each metal is used.[5][20][21] The alkali metals have the lowest first ionisation energies in their respective periods of the periodic table[7] because of their low effective nuclear charge[5] and the ability to attain a noble gas configuration by losing just one electron.[5] Not only do the alkali metals react with water, but also with proton donors like alcohols and phenols, gaseous ammonia, and alkynes, the last demonstrating the phenomenal degree of their reactivity. Their great power as reducing agents makes them very useful in liberating other metals from their oxides or halides.[6]:76
The second ionisation energy of all of the alkali metals is very high[5][7] as it is in a full shell that is also closer to the nucleus;[5] thus, they almost always lose a single electron, forming cations.[6]:28 The alkalides are an exception: they are unstable compounds which contain alkali metals in a −1 oxidation state, which is very unusual as before the discovery of the alkalides, the alkali metals were not expected to be able to form anions and were thought to be able to appear in salts only as cations. The alkalide anions have filled s-subshells, which gives them enough stability to exist. All the stable alkali metals except lithium are known to be able to form alkalides,[22][23][24] and the alkalides have much theoretical interest due to their unusual stoichiometry and low ionisation potentials. Alkalides are chemically similar to the electrides, which are salts with trapped electrons acting as anions.[25] A particularly striking example of an alkalide is "inverse sodium hydride", H+Na− (both ions being complexed), as opposed to the usual sodium hydride, Na+H−:[26] it is unstable in isolation, due to its high energy resulting from the displacement of two electrons from hydrogen to sodium, although several derivatives are predicted to be metastable or stable.[26][27]
In aqueous solution, the alkali metal ions form aqua ions of the formula [M(H2O)n]+, where n is the solvation number. Their coordination numbers and shapes agree well with those expected from their ionic radii. In aqueous solution the water molecules directly attached to the metal ion are said to belong to the first coordination sphere, also known as the first, or primary, solvation shell. The bond between a water molecule and the metal ion is a dative covalent bond, with the oxygen atom donating both electrons to the bond. Each coordinated water molecule may be attached by hydrogen bonds to other water molecules. The latter are said to reside in the second coordination sphere. However, for the alkali metal cations, the second coordination sphere is not well-defined as the +1 charge on the cation is not high enough to polarise the water molecules in the primary solvation shell enough for them to form strong hydrogen bonds with those in the second coordination sphere, producing a more stable entity.[28][29]:25 The solvation number for Li+ has been experimentally determined to be 4, forming the tetrahedral [Li(H2O)4]+: while solvation numbers of 3 to 6 have been found for lithium aqua ions, solvation numbers less than 4 may be the result of the formation of contact ion pairs, and the higher solvation numbers may be interpreted in terms of water molecules that approach [Li(H2O)4]+ through a face of the tetrahedron, though molecular dynamic simulations may indicate the existence of an octahedral hexaaqua ion. There are also probably six water molecules in the primary solvation sphere of the sodium ion, forming the octahedral [Na(H2O)6]+ ion.[8][29]:126–127 While it was previously thought that the heavier alkali metals also formed octahedral hexaaqua ions, it has since been found that potassium and rubidium probably form the [K(H2O)8]+ and [Rb(H2O)8]+ ions, which have the square antiprismatic structure, and that caesium forms the 12-coordinate [Cs(H2O)12]+ ion.[30]
Lithium
The chemistry of lithium shows several differences from that of the rest of the group as the small Li+ cation polarises anions and gives its compounds a more covalent character.[5] Lithium and magnesium have a diagonal relationship due to their similar atomic radii,[5] so that they show some similarities. For example, lithium forms a stable nitride, a property common among all the alkaline earth metals (magnesium's group) but unique among the alkali metals.[31] In addition, among their respective groups, only lithium and magnesium form organometallic compounds with significant covalent character (e.g. LiMe and MgMe2).[32]
Lithium fluoride is the only alkali metal halide that is poorly soluble in water,[5] and lithium hydroxide is the only alkali metal hydroxide that is not deliquescent.[5] Conversely, lithium perchlorate and other lithium salts with large anions that cannot be polarised are much more stable than the analogous compounds of the other alkali metals, probably because Li+ has a high solvation energy.[6]:76 This effect also means that most simple lithium salts are commonly encountered in hydrated form, because the anhydrous forms are extremely hygroscopic: this allows salts like lithium chloride and lithium bromide to be used in dehumidifiers and air-conditioners.[6]:76
Francium
Francium is also predicted to show some differences due to its high atomic weight, causing its electrons to travel at considerable fractions of the speed of light and thus making relativistic effects more prominent. In contrast to the trend of decreasing electronegativities and ionisation energies of the alkali metals, francium's electronegativity and ionisation energy are predicted to be higher than caesium's due to the relativistic stabilisation of the 7s electrons; also, its atomic radius is expected to be abnormally low. Thus, contrary to expectation, caesium is the most reactive of the alkali metals, not francium.[12][33]:1729[34] All known physical properties of francium also deviate from the clear trends going from lithium to caesium, such as the first ionisation energy, electron affinity, and anion polarisability, though due to the paucity of known data about francium many sources give extrapolated values, ignoring that relativistic effects make the trend from lithium to caesium become inapplicable at francium.[34] Some of the few properties of francium that have been predicted taking relativity into account are the electron affinity (47.2 kJ/mol)[35] and the enthalpy of dissociation of the Fr2 molecule (42.1 kJ/mol).[36] The CsFr molecule is polarised as Cs+Fr−, showing that the 7s subshell of francium is much more strongly affected by relativistic effects than the 6s subshell of caesium.[34] Additionally, francium superoxide (FrO2) is expected to have significant covalent character, unlike the other alkali metal superoxides, because of bonding contributions from the 6p electrons of francium.[34]
Nuclear
| Z |
Alkali metal |
Stable |
Decays |
unstable: italics odd–odd isotopes coloured pink | ||
|---|---|---|---|---|---|---|
| 3 | lithium | 2 | — | 7 Li | 6 Li | |
| 11 | sodium | 1 | — | 23 Na | ||
| 19 | potassium | 2 | 1 | 39 K | 41 K | 40 K |
| 37 | rubidium | 1 | 1 | 85 Rb | 87 Rb | |
| 55 | caesium | 1 | — | 133 Cs | ||
| 87 | francium | — | — | No primordial isotopes (223 Fr is a radiogenic nuclide) | ||
| Radioactive: 40K, t1/2 1.25 × 109 years; 87Rb, t1/2 4.9 × 1010 years; 223Fr, t1/2 22.0 min. | ||||||
All the alkali metals have odd atomic numbers; hence, their isotopes must be either odd–odd (both proton and neutron number are odd) or odd–even (proton number is odd, but neutron number is even). Odd–odd nuclei have even mass numbers, whereas odd–even nuclei have odd mass numbers. Odd–odd primordial nuclides are rare because most odd–odd nuclei are highly unstable with respect to beta decay, because the decay products are even–even, and are therefore more strongly bound, due to nuclear pairing effects.[37]
Due to the great rarity of odd–odd nuclei, almost all the primordial isotopes of the alkali metals are odd–even (the exceptions being the light stable isotope lithium-6 and the long-lived radioisotope potassium-40). For a given odd mass number, there can be only a single beta-stable nuclide, since there is not a difference in binding energy between even–odd and odd–even comparable to that between even–even and odd–odd, leaving other nuclides of the same mass number (isobars) free to beta decay toward the lowest-mass nuclide. An effect of the instability of an odd number of either type of nucleons is that odd-numbered elements, such as the alkali metals, tend to have fewer stable isotopes than even-numbered elements. Of the 26 monoisotopic elements that have only a single stable isotope, all but one have an odd atomic number and all but one also have an even number of neutrons. Beryllium is the single exception to both rules, due to its low atomic number.[37]
All of the alkali metals except lithium and caesium have at least one naturally occurring radioisotope: sodium-22 and sodium-24 are trace radioisotopes produced cosmogenically,[38] potassium-40 and rubidium-87 have very long half-lives and thus occur naturally,[39] and all isotopes of francium are radioactive.[39] Caesium was also thought to be radioactive in the early 20th century,[40][41] although it has no naturally occurring radioisotopes.[39] (Francium had not been discovered yet at that time.) The natural long-lived radioisotope of potassium, potassium-40, makes up about 0.012% of natural potassium,[42] and thus natural potassium is weakly radioactive. This natural radioactivity became a basis for a mistaken claim of the discovery for element 87 (the next alkali metal after caesium) in 1925.[43][44] Natural rubidium is similarly slightly radioactive, with 27.83% being the long-lived radioisotope rubidium-87.[6]:74
Caesium-137, with a half-life of 30.17 years, is one of the two principal medium-lived fission products, along with strontium-90, which are responsible for most of the radioactivity of spent nuclear fuel after several years of cooling, up to several hundred years after use. It constitutes most of the radioactivity still left from the Chernobyl accident. Caesium-137 undergoes high-energy beta decay and eventually becomes stable barium-137. It is a strong emitter of gamma radiation. Caesium-137 has a very low rate of neutron capture and cannot be feasibly disposed of in this way, but must be allowed to decay.[45] Caesium-137 has been used as a tracer in hydrologic studies, analogous to the use of tritium.[46] Small amounts of caesium-134 and caesium-137 were released into the environment during nearly all nuclear weapon tests and some nuclear accidents, most notably the Goiânia accident and the Chernobyl disaster. As of 2005, caesium-137 is the principal source of radiation in the zone of alienation around the Chernobyl nuclear power plant.[47] Its chemical properties as one of the alkali metals make it one of most problematic of the short-to-medium-lifetime fission products because it easily moves and spreads in nature due to the high water solubility of its salts, and is taken up by the body, which mistakes it for its essential congeners sodium and potassium.[48]:114
Periodic trends
The alkali metals are more similar to each other than the elements in any other group are to each other.[5] For instance, when moving down the table, all known alkali metals show increasing atomic radius,[16] decreasing electronegativity,[16] increasing reactivity,[5] and decreasing melting and boiling points[16] as well as heats of fusion and vaporisation.[6]:75 In general, their densities increase when moving down the table, with the exception that potassium is less dense than sodium.[16]
Atomic and ionic radii
The atomic radii of the alkali metals increase going down the group.[16] Because of the shielding effect, when an atom has more than one electron shell, each electron feels electric repulsion from the other electrons as well as electric attraction from the nucleus.[49] In the alkali metals, the outermost electron only feels a net charge of +1, as some of the nuclear charge (which is equal to the atomic number) is cancelled by the inner electrons; the number of inner electrons of an alkali metal is always one less than the nuclear charge. Therefore, the only factor which affects the atomic radius of the alkali metals is the number of electron shells. Since this number increases down the group, the atomic radius must also increase down the group.[16]
The ionic radii of the alkali metals are much smaller than their atomic radii. This is because the outermost electron of the alkali metals is in a different electron shell than the inner electrons, and thus when it is removed the resulting atom has one fewer electron shell and is smaller. Additionally, the effective nuclear charge has increased, and thus the electrons are attracted more strongly towards the nucleus and the ionic radius decreases.[5]
First ionisation energy

The first ionisation energy of an element or molecule is the energy required to move the most loosely held electron from one mole of gaseous atoms of the element or molecules to form one mole of gaseous ions with electric charge +1. The factors affecting the first ionisation energy are the nuclear charge, the amount of shielding by the inner electrons and the distance from the most loosely held electron from the nucleus, which is always an outer electron in main group elements. The first two factors change the effective nuclear charge the most loosely held electron feels. Since the outermost electron of alkali metals always feels the same effective nuclear charge (+1), the only factor which affects the first ionisation energy is the distance from the outermost electron to the nucleus. Since this distance increases down the group, the outermost electron feels less attraction from the nucleus and thus the first ionisation energy decreases.[16] (This trend is broken in francium due to the relativistic stabilisation and contraction of the 7s orbital, bringing francium's valence electron closer to the nucleus than would be expected from non-relativistic calculations. This makes francium's outermost electron feel more attraction from the nucleus, increasing its first ionisation energy slightly beyond that of caesium.)[33]:1729
The second ionisation energy of the alkali metals is much higher than the first as the second-most loosely held electron is part of a fully filled electron shell and is thus difficult to remove.[5]
Reactivity
The reactivities of the alkali metals increase going down the group. This is the result of a combination of two factors: the first ionisation energies and atomisation energies of the alkali metals. Because the first ionisation energy of the alkali metals decreases down the group, it is easier for the outermost electron to be removed from the atom and participate in chemical reactions, thus increasing reactivity down the group. The atomisation energy measures the strength of the metallic bond of an element, which falls down the group as the atoms increase in radius and thus the metallic bond must increase in length, making the delocalised electrons further away from the attraction of the nuclei of the heavier alkali metals. Adding the atomisation and first ionisation energies gives a quantity closely related to (but not equal to) the activation energy of the reaction of an alkali metal with another substance. This quantity decreases going down the group, and so does the activation energy; thus, chemical reactions can occur faster and the reactivity increases down the group.[50]
Electronegativity

Electronegativity is a chemical property that describes the tendency of an atom or a functional group to attract electrons (or electron density) towards itself.[51] If the bond between sodium and chlorine in sodium chloride were covalent, the pair of shared electrons would be attracted to the chlorine because the effective nuclear charge on the outer electrons is +7 in chlorine but is only +1 in sodium. The electron pair is attracted so close to the chlorine atom that they are practically transferred to the chlorine atom (an ionic bond). However, if the sodium atom was replaced by a lithium atom, the electrons will not be attracted as close to the chlorine atom as before because the lithium atom is smaller, making the electron pair more strongly attracted to the closer effective nuclear charge from lithium. Hence, the larger alkali metal atoms (further down the group) will be less electronegative as the bonding pair is less strongly attracted towards them. As mentioned previously, francium is expected to be an exception.[16]
Because of the higher electronegativity of lithium, some of its compounds have a more covalent character. For example, lithium iodide (LiI) will dissolve in organic solvents, a property of most covalent compounds.[16] Lithium fluoride (LiF) is the only alkali halide that is not soluble in water,[5] and lithium hydroxide (LiOH) is the only alkali metal hydroxide that is not deliquescent.[5]
Melting and boiling points
The melting point of a substance is the point where it changes state from solid to liquid while the boiling point of a substance (in liquid state) is the point where the vapour pressure of the liquid equals the environmental pressure surrounding the liquid[52][53] and all the liquid changes state to gas. As a metal is heated to its melting point, the metallic bonds keeping the atoms in place weaken so that the atoms can move around, and the metallic bonds eventually break completely at the metal's boiling point.[16][54] Therefore, the falling melting and boiling points of the alkali metals indicate that the strength of the metallic bonds of the alkali metals decreases down the group.[16] This is because metal atoms are held together by the electromagnetic attraction from the positive ions to the delocalised electrons.[16][54] As the atoms increase in size going down the group (because their atomic radius increases), the nuclei of the ions move further away from the delocalised electrons and hence the metallic bond becomes weaker so that the metal can more easily melt and boil, thus lowering the melting and boiling points.[16] (The increased nuclear charge is not a relevant factor due to the shielding effect.)[16]
Density
The alkali metals all have the same crystal structure (body-centred cubic)[6] and thus the only relevant factors are the number of atoms that can fit into a certain volume and the mass of one of the atoms, since density is defined as mass per unit volume. The first factor depends on the volume of the atom and thus the atomic radius, which increases going down the group; thus, the volume of an alkali metal atom increases going down the group. The mass of an alkali metal atom also increases going down the group. Thus, the trend for the densities of the alkali metals depends on their atomic weights and atomic radii; if figures for these two factors are known, the ratios between the densities of the alkali metals can then be calculated. The resultant trend is that the densities of the alkali metals increase down the table, with an exception at potassium. Due to having the lowest atomic weight of all the elements in their period and having the largest atomic radius for their periods, the alkali metals are the least dense metals in the periodic table.[16] Lithium, sodium, and potassium are the only three metals in the periodic table that are less dense than water:[5] in fact, lithium is the least dense known solid at room temperature.[6]:75
Compounds
The alkali metals form complete series of compounds with all usually encountered anions, which well illustrate group trends. These compounds can be described as involving the alkali metals losing electrons to acceptor species and forming monopositive ions.[6]:79 This description is most accurate for alkali halides and becomes less and less accurate as cationic and anionic charge increase, and as the anion becomes larger and more polarisable. For instance, ionic bonding gives way to metallic bonding along the series NaCl, Na2O, Na2S, Na3P, Na3As, Na3Sb, Na3Bi, Na.[6]:81
Hydroxides
|
|

All the alkali metals react vigorously or explosively with cold water, producing an aqueous solution of a strongly basic alkali metal hydroxide and releasing hydrogen gas.[50] This reaction becomes more vigorous going down the group: lithium reacts steadily with effervescence, but sodium and potassium can ignite and rubidium and caesium sink in water and generate hydrogen gas so rapidly that shock waves form in the water that may shatter glass containers.[5] When an alkali metal is dropped into water, it produces an explosion, of which there are two separate stages. The metal reacts with the water first, breaking the hydrogen bonds in the water and producing hydrogen gas; this takes place faster for the more reactive heavier alkali metals. Second, the heat generated by the first part of the reaction often ignites the hydrogen gas, causing it to burn explosively into the surrounding air. This secondary hydrogen gas explosion produces the visible flame above the bowl of water, lake or other body of water, not the initial reaction of the metal with water (which tends to happen mostly under water).[20] The alkali metal hydroxides are the most basic known hydroxides.[6]:87
Recent research has suggested that the explosive behavior of alkali metals in water is driven by a Coulomb explosion rather than solely by rapid generation of hydrogen itself.[55] All alkali metals melt as a part of the reaction with water. Water molecules ionise the bare metallic surface of the liquid metal, leaving a positively charged metal surface and negatively charged water ions. The attraction between the charged metal and water ions will rapidly increase the surface area, causing an exponential increase of ionisation. When the repulsive forces within the liquid metal surface exceeds the forces of the surface tension, it vigorously explodes.[55]
The hydroxides themselves are the most basic hydroxides known, reacting with acids to give salts and with alcohols to give oligomeric alkoxides. They easily react with carbon dioxide to form carbonates or bicarbonates, or with hydrogen sulfide to form sulfides or bisulfides, and may be used to separate thiols from petroleum. They react with amphoteric oxides: for example, the oxides of aluminium, zinc, tin, and lead react with the alkali metal hydroxides to give aluminates, zincates, stannates, and plumbates. Silicon dioxide is acidic, and thus the alkali metal hydroxides can also attack silicate glass.[6]:87
Intermetallic compounds

The alkali metals form many intermetallic compounds with each other and the elements from groups 2 to 13 in the periodic table of varying stoichiometries,[6]:81 such as the sodium amalgams with mercury, including Na5Hg8 and Na3Hg.[56] Some of these have ionic characteristics: taking the alloys with gold, the most electronegative of metals, as an example, NaAu and KAu are metallic, but RbAu and CsAu are semiconductors.[6]:81 NaK is an alloy of sodium and potassium that is very useful because it is liquid at room temperature, although precautions must be taken due to its extreme reactivity towards water and air. The eutectic mixture melts at −12.6 °C.[57] An alloy of 41% caesium, 47% sodium, and 12% potassium has the lowest known melting point of any metal or alloy, −78 °C.[58]
Compounds with the group 13 elements
The intermetallic compounds of the alkali metals with the heavier group 13 elements (aluminium, gallium, indium, and thallium), such as NaTl, are poor conductors or semiconductors, unlike the normal alloys with the preceding elements, implying that the alkali metal involved has lost an electron to the Zintl anions involved.[59] Nevertheless, while the elements in group 14 and beyond tend to form discrete anionic clusters, group 13 elements tend to form polymeric ions with the alkali metal cations located between the giant ionic lattice. For example, NaTl consists of a polymeric anion (—Tl−—)n with a covalent diamond cubic structure with Na+ ions located between the anionic lattice. The larger alkali metals cannot fit similarly into an anionic lattice and tend to force the heavier group 13 elements to form anionic clusters.[60]
Boron is a special case, being the only nonmetal in group 13. The alkali metal borides tend to be boron-rich, involving appreciable boron–boron bonding involving deltahedral structures,[6]:147–8 and are thermally unstable due to the alkali metals having a very high vapour pressure at elevated temperatures. This makes direct synthesis problematic because the alkali metals do not react with boron below 700 °C, and thus this must be accomplished in sealed containers with the alkali metal in excess. Furthermore, exceptionally in this group, reactivity with boron decreases down the group: lithium reacts completely at 700 °C, but sodium at 900 °C and potassium not until 1200 °C, and the reaction is instantaneous for lithium but takes hours for potassium. Rubidium and caesium borides have not even been characterised. Various phases are known, such as LiB10, NaB6, NaB15, and KB6.[61][62] Under high pressure the boron–boron bonding in the lithium borides changes from following Wade's rules to forming Zintl anions like the rest of group 13.[63]
Compounds with the group 14 elements


Lithium and sodium react with carbon to form acetylides, Li2C2 and Na2C2, which can also be obtained by reaction of the metal with acetylene. Potassium, rubidium, and caesium react with graphite; their atoms are intercalated between the hexagonal graphite layers, forming graphite intercalation compounds of formulae MC60 (dark grey, almost black), MC48 (dark grey, almost black), MC36 (blue), MC24 (steel blue), and MC8 (bronze) (M = K, Rb, or Cs). These compounds are over 200 times more electrically conductive than pure graphite, suggesting that the valence electron of the alkali metal is transferred to the graphite layers (e.g. M+
C−
8).[8] Upon heating of KC8, the elimination of potassium atoms results in the conversion in sequence to KC24, KC36, KC48 and finally KC60. KC8 is a very strong reducing agent and is pyrophoric and explodes on contact with water.[64][65] While the larger alkali metals (K, Rb, and Cs) initially form MC8, the smaller ones initially form MC6, and indeed they require reaction of the metals with graphite at high temperatures around 500 °C to form.[66] Apart from this, the alkali metals are such strong reducing agents that they can even reduce buckminsterfullerene to produce solid fullerides MnC60; sodium, potassium, rubidium, and caesium can form fullerides where n = 2, 3, 4, or 6, and rubidium and caesium additionally can achieve n = 1.[6]:285
When the alkali metals react with the heavier elements in the carbon group (silicon, germanium, tin, and lead), ionic substances with cage-like structures are formed, such as the silicides M4Si4 (M = K, Rb, or Cs), which contains M+ and tetrahedral Si4−
4 ions.[8] The chemistry of alkali metal germanides, involving the germanide ion Ge4− and other cluster (Zintl) ions such as Ge2−
4, Ge4−
9, Ge2−
9, and [(Ge9)2]6−, is largely analogous to that of the corresponding silicides.[6]:393 Alkali metal stannides are mostly ionic, sometimes with the stannide ion (Sn4−),[60] and sometimes with more complex Zintl ions such as Sn4−
9, which appears in tetrapotassium nonastannide (K4Sn9).[67] The monatomic plumbide ion (Pb4−) is unknown, and indeed its formation is predicted to be energetically unfavourable; alkali metal plumbides have complex Zintl ions, such as Pb4−
9. These alkali metal germanides, stannides, and plumbides may be produced by reducing germanium, tin, and lead with sodium metal in liquid ammonia.[6]:394
Nitrides and pnictides

Lithium, the lightest of the alkali metals, is the only alkali metal which reacts with nitrogen at standard conditions, and its nitride is the only stable alkali metal nitride. Nitrogen is an unreactive gas because breaking the strong triple bond in the dinitrogen molecule (N2) requires a lot of energy. The formation of an alkali metal nitride would consume the ionisation energy of the alkali metal (forming M+ ions), the energy required to break the triple bond in N2 and the formation of N3− ions, and all the energy released from the formation of an alkali metal nitride is from the lattice energy of the alkali metal nitride. The lattice energy is maximised with small, highly charged ions; the alkali metals do not form highly charged ions, only forming ions with a charge of +1, so only lithium, the smallest alkali metal, can release enough lattice energy to make the reaction with nitrogen exothermic, forming lithium nitride. The reactions of the other alkali metals with nitrogen would not release enough lattice energy and would thus be endothermic, so they do not form nitrides at standard conditions.[31] Sodium nitride (Na3N) and potassium nitride (K3N), while existing, are extremely unstable, being prone to decomposing back into their constituent elements, and cannot be produced by reacting the elements with each other at standard conditions.[69][70] Steric hindrance forbids the existence of rubidium or caesium nitride.[6]:417 However, sodium and potassium form colourless azide salts involving the linear N−
3 anion; due to the large size of the alkali metal cations, they are thermally stable enough to be able to melt before decomposing.[6]:417
All the alkali metals react readily with phosphorus and arsenic to form phosphides and arsenides with the formula M3Pn (where M represents an alkali metal and Pn represents a pnictogen – phosphorus, arsenic, antimony, or bismuth). This is due to the greater size of the P3− and As3− ions, so that less lattice energy needs to be released for the salts to form.[8] These are not the only phosphides and arsenides of the alkali metals: for example, potassium has nine different known phosphides, with formulae K3P, K4P3, K5P4, KP, K4P6, K3P7, K3P11, KP10.3, and KP15.[71] While most metals form arsenides, only the alkali and alkaline earth metals form mostly ionic arsenides. The structure of Na3As is complex with unusually short Na–Na distances of 328–330 pm which are shorter than in sodium metal, and this indicates that even with these electropositive metals the bonding cannot be straightforwardly ionic.[6] Other alkali metal arsenides not conforming to the formula M3As are known, such as LiAs, which has a metallic lustre and electrical conductivity indicating the presence of some metallic bonding.[6] The antimonides are unstable and reactive as the Sb3− ion is a strong reducing agent; reaction of them with acids form the toxic and unstable gas stibine (SbH3).[72] Indeed, they have some metallic properties, and the alkali metal antimonides of stoichiometry MSb involve antimony atoms bonded in a spiral Zintl structure.[73] Bismuthides are not even wholly ionic; they are intermetallic compounds containing partially metallic and partially ionic bonds.[74]
Oxides and chalcogenides

9O
2 cluster, composed of two regular octahedra connected to each other by one face
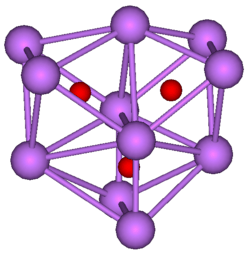
11O
3 cluster, composed of three regular octahedra where each octahedron is connected to both of the others by one face each. All three octahedra have one edge in common.
All the alkali metals react vigorously with oxygen at standard conditions. They form various types of oxides, such as simple oxides (containing the O2− ion), peroxides (containing the O2−
2 ion, where there is a single bond between the two oxygen atoms), superoxides (containing the O−
2 ion), and many others. Lithium burns in air to form lithium oxide, but sodium reacts with oxygen to form a mixture of sodium oxide and sodium peroxide. Potassium forms a mixture of potassium peroxide and potassium superoxide, while rubidium and caesium form the superoxide exclusively. Their reactivity increases going down the group: while lithium, sodium and potassium merely burn in air, rubidium and caesium are pyrophoric (spontaneously catch fire in air).[31]
The smaller alkali metals tend to polarise the larger anions (the peroxide and superoxide) due to their small size. This attracts the electrons in the more complex anions towards one of its constituent oxygen atoms, forming an oxide ion and an oxygen atom. This causes lithium to form the oxide exclusively on reaction with oxygen at room temperature. This effect becomes drastically weaker for the larger sodium and potassium, allowing them to form the less stable peroxides. Rubidium and caesium, at the bottom of the group, are so large that even the least stable superoxides can form. Because the superoxide releases the most energy when formed, the superoxide is preferentially formed for the larger alkali metals where the more complex anions are not polarised. (The oxides and peroxides for these alkali metals do exist, but do not form upon direct reaction of the metal with oxygen at standard conditions.)[31] In addition, the small size of the Li+ and O2− ions contributes to their forming a stable ionic lattice structure. Under controlled conditions, however, all the alkali metals, with the exception of francium, are known to form their oxides, peroxides, and superoxides. The alkali metal peroxides and superoxides are powerful oxidising agents. Sodium peroxide and potassium superoxide react with carbon dioxide to form the alkali metal carbonate and oxygen gas, which allows them to be used in submarine air purifiers; the presence of water vapour, naturally present in breath, makes the removal of carbon dioxide by potassium superoxide even more efficient.[8][75] All the stable alkali metals except lithium can form red ozonides (MO3) through low-temperature reaction of the powdered anhydrous hydroxide with ozone: the ozonides may be then extracted using liquid ammonia. They slowly decompose at standard conditions to the superoxides and oxygen, and hydrolyse immediately to the hydroxides when in contact with water.[6]:85 Potassium, rubidium, and caesium also form sesquioxides M2O3, which may be better considered peroxide disuperoxides, [(M+
)
4(O2−
2)(O−
2)
2].[6]:85
Rubidium and caesium can form a great variety of suboxides with the metals in formal oxidation states below +1.[6]:85 Rubidium can form Rb6O and Rb9O2 (copper-coloured) upon oxidation in air, while caesium forms an immense variety of oxides, such as the ozonide CsO3[76][77] and several brightly coloured suboxides,[78] such as Cs7O (bronze), Cs4O (red-violet), Cs11O3 (violet), Cs3O (dark green),[79] CsO, Cs3O2,[80] as well as Cs7O2.[81][82] The last of these may be heated under vacuum to generate Cs2O.[83]
The alkali metals can also react analogously with the heavier chalcogens (sulfur, selenium, tellurium, and polonium), and all the alkali metal chalcogenides are known (with the exception of francium's). Reaction with an excess of the chalcogen can similarly result in lower chalcogenides, with chalcogen ions containing chains of the chalcogen atoms in question. For example, sodium can react with sulfur to form the sulfide (Na2S) and various polysulfides with the formula Na2Sx (x from 2 to 6), containing the S2−
x ions.[8] Due to the basicity of the Se2− and Te2− ions, the alkali metal selenides and tellurides are alkaline in solution; when reacted directly with selenium and tellurium, alkali metal polyselenides and polytellurides are formed along with the selenides and tellurides with the Se2−
x and Te2−
x ions.[84] They may be obtained directly from the elements in liquid ammonia or when air is not present, and are colourless, water-soluble compounds that air oxidises quickly back to selenium or tellurium.[6]:766 The alkali metal polonides are all ionic compounds containing the Po2− ion; they are very chemically stable and can be produced by direct reaction of the elements at around 300–400 °C.[6]:766[85][86]
Halides, hydrides, and pseudohalides
The alkali metals are among the most electropositive elements on the periodic table and thus tend to bond ionically to the most electronegative elements on the periodic table, the halogens (fluorine, chlorine, bromine, iodine, and astatine), forming salts known as the alkali metal halides. The reaction is very vigorous and can sometimes result in explosions.[6]:76 All twenty stable alkali metal halides are known; the unstable ones are not known, with the exception of sodium astatide, because of the great instability and rarity of astatine and francium. The most well-known of the twenty is certainly sodium chloride, otherwise known as common salt. All of the stable alkali metal halides have the formula MX where M is an alkali metal and X is a halogen. They are all white ionic crystalline solids that have high melting points.[5][31] All the alkali metal halides are soluble in water except for lithium fluoride (LiF), which is insoluble in water due to its very high lattice enthalpy. The high lattice enthalpy of lithium fluoride is due to the small sizes of the Li+ and F− ions, causing the electrostatic interactions between them to be strong:[5] a similar effect occurs for magnesium fluoride, consistent with the diagonal relationship between lithium and magnesium.[6]:76
The alkali metals also react similarly with hydrogen to form ionic alkali metal hydrides, where the hydride anion acts as a pseudohalide: these are often used as reducing agents, producing hydrides, complex metal hydrides, or hydrogen gas.[6]:83[8] Other pseudohalides are also known, notably the cyanides. These are isostructural to the respective halides except for lithium cyanide, indicating that the cyanide ions may rotate freely.[6]:322 Ternary alkali metal halide oxides, such as Na3ClO, K3BrO (yellow), Na4Br2O, Na4I2O, and K4Br2O, are also known.[6]:83 The polyhalides are rather unstable, although those of rubidium and caesium are greatly stabilised by the feeble polarising power of these extremely large cations.[6]:835
Coordination complexes


Alkali metal cations do not usually form coordination complexes with simple Lewis bases due to their low charge of just +1 and their relatively large size; thus the Li+ ion forms most complexes and the heavier alkali metal ions form less and less (though exceptions occur for weak complexes).[6]:90 Lithium in particular has a very rich coordination chemistry in which it exhibits coordination numbers from 1 to 12, although octahedral hexacoordination is its preferred mode.[6]:90–1 In aqueous solution, the alkali metal ions exist as octahedral hexahydrate complexes ([M(H2O)6)]+), with the exception of the lithium ion, which due to its small size forms tetrahedral tetrahydrate complexes ([Li(H2O)4)]+); the alkali metals form these complexes because their ions are attracted by electrostatic forces of attraction to the polar water molecules. Because of this, anhydrous salts containing alkali metal cations are often used as desiccants.[8] Alkali metals also readily form complexes with crown ethers (e.g. 12-crown-4 for Li+, 15-crown-5 for Na+, 18-crown-6 for K+, and 21-crown-7 for Rb+) and cryptands due to electrostatic attraction.[8]
Ammonia solutions
The alkali metals dissolve slowly in liquid ammonia, forming ammoniacal solutions of solvated M+ and e−, which react to form hydrogen gas and the alkali metal amide (MNH2, where M represents an alkali metal): this was first noted by Humphry Davy in 1809 and rediscovered by W. Weyl in 1864. The process may be speeded up by a catalyst. Similar solutions are formed by the heavy divalent alkaline earth metals calcium, strontium, barium, as well as the divalent lanthanides, europium and ytterbium. The amide salt is quite insoluble and readily precipitates out of solution, leaving intensely coloured ammonia solutions of the alkali metals. In 1907, Charles Krause identified the colour as being due to the presence of solvated electrons, which contribute to the high electrical conductivity of these solutions. At low concentrations (below 3 M), the solution is dark blue and has ten times the conductivity of aqueous sodium chloride; at higher concentrations (above 3 M), the solution is copper-coloured and has approximately the conductivity of liquid metals like mercury.[6][8][88] In addition to the alkali metal amide salt and solvated electrons, such ammonia solutions also contain the alkali metal cation (M+), the neutral alkali metal atom (M), diatomic alkali metal molecules (M2) and alkali metal anions (M−). These are unstable and eventually become the more thermodynamically stable alkali metal amide and hydrogen gas. Solvated electrons are powerful reducing agents and are often used in chemical synthesis.[8]
Organometallic
Organolithium


Being the smallest alkali metal, lithium forms the widest variety of and most stable organometallic compounds, which are bonded covalently. Organolithium compounds are electrically non-conducting volatile solids or liquids that melt at low temperatures, and tend to form oligomers with the structure (RLi)x where R is the organic group. As the electropositive nature of lithium puts most of the charge density of the bond on the carbon atom, effectively creating a carbanion, organolithium compounds are extremely powerful bases and nucleophiles. For use as bases, butyllithiums are often used and are commercially available. An example of an organolithium compound is methyllithium ((CH3Li)x), which exists in tetrameric (x = 4, tetrahedral) and hexameric (x = 6, octahedral) forms.[8][92] Organolithium compounds, especially n-butyllithium, are useful reagents in organic synthesis, as might be expected given lithium's diagonal relationship with magnesium, which plays an important role in the Grignard reaction.[6]:102 For example, alkyllithiums and aryllithiums may be used to synthesise aldehydes and ketones by reaction with metal carbonyls. The reaction with nickel tetracarbonyl, for example, proceeds through an unstable acyl nickel carbonyl complex which then undergoes electrophilic substitution to give the desired aldehyde (using H+ as the electrophile) or ketone (using an alkyl halide) product.[6]:105
- LiR + [Ni(CO)4] Li+[RCONi(CO)3]−
- Li+[RCONi(CO)3]− Li+ + RCHO + [(solvent)Ni(CO)3]
- Li+[RCONi(CO)3]− Li+ + R'COR + [(solvent)Ni(CO)3]
Alkyllithiums and aryllithiums may also react with N,N-disubstituted amides to give aldehydes and ketones, and symmetrical ketones by reacting with carbon monoxide. They thermally decompose to eliminate a β-hydrogen, producing alkenes and lithium hydride: another route is the reaction of ethers with alkyl- and aryllithiums that act as strong bases.[6]:105 In non-polar solvents, aryllithiums react as the carbanions they effectively are, turning carbon dioxide to aromatic carboxylic acids (ArCO2H) and aryl ketones to tertiary carbinols (Ar'2C(Ar)OH). Finally, they may be used to synthesise other organometallic compounds through metal-halogen exchange.[6]:106
Heavier alkali metals
Unlike the organolithium compounds, the organometallic compounds of the heavier alkali metals are predominantly ionic. The application of organosodium compounds in chemistry is limited in part due to competition from organolithium compounds, which are commercially available and exhibit more convenient reactivity. The principal organosodium compound of commercial importance is sodium cyclopentadienide. Sodium tetraphenylborate can also be classified as an organosodium compound since in the solid state sodium is bound to the aryl groups. Organometallic compounds of the higher alkali metals are even more reactive than organosodium compounds and of limited utility. A notable reagent is Schlosser's base, a mixture of n-butyllithium and potassium tert-butoxide. This reagent reacts with propene to form the compound allylpotassium (KCH2CHCH2). cis-2-Butene and trans-2-butene equilibrate when in contact with alkali metals. Whereas isomerisation is fast with lithium and sodium, it is slow with the heavier alkali metals. The heavier alkali metals also favour the sterically congested conformation.[93] Several crystal structures of organopotassium compounds have been reported, establishing that they, like the sodium compounds, are polymeric.[94] Organosodium, organopotassium, organorubidium and organocaesium compounds are all mostly ionic and are insoluble (or nearly so) in nonpolar solvents.[8]
Alkyl and aryl derivatives of sodium and potassium tend to react with air. They cause the cleavage of ethers, generating alkoxides. Unlike alkyllithium compounds, alkylsodiums and alkylpotassiums cannot be made by reacting the metals with alkyl halides because Wurtz coupling occurs:[73]:265
- RM + R'X → R–R' + MX
As such, they have to be made by reacting alkylmercury compounds with sodium or potassium metal in inert hydrocarbon solvents. While methylsodium forms tetramers like methyllithium, methylpotassium is more ionic and has the nickel arsenide structure with discrete methyl anions and potassium cations.[73]:265
The alkali metals and their hydrides react with acidic hydrocarbons, for example cyclopentadienes and terminal alkynes, to give salts. Liquid ammonia, ether, or hydrocarbon solvents are used, the most common of which being tetrahydrofuran. The most important of these compounds is sodium cyclopentadienide, NaC5H5, an important precursor to many transition metal cyclopentadienyl derivatives.[73]:265 Similarly, the alkali metals react with cyclooctatetraene in tetrahydrofuran to give alkali metal cyclooctatetraenides; for example, dipotassium cyclooctatetraenide (K2C8H8) is an important precursor to many metal cyclooctatetraenyl derivatives, such as uranocene.[73]:266 The large and very weakly polarising alkali metal cations can stabilise large, aromatic, polarisable radical anions, such as the dark-green sodium naphthalenide, Na+[C10H8•]−, a strong reducing agent.[73]:266
Extensions

Although francium is the heaviest alkali metal that has been discovered, there has been some theoretical work predicting the physical and chemical characteristics of the hypothetical heavier alkali metals. Being the first period 8 element, the undiscovered element ununennium (element 119) is predicted to be the next alkali metal after francium and behave much like their lighter congeners; however, it is also predicted to differ from the lighter alkali metals in some properties.[33]:1729–1730 Its chemistry is predicted to be closer to that of potassium[96] or rubidium[33]:1729–1730 instead of caesium or francium. This is unusual as periodic trends, ignoring relativistic effects would predict ununennium to be even more reactive than caesium and francium. This lowered reactivity is due to the relativistic stabilisation of ununennium's valence electron, increasing ununennium's first ionisation energy and decreasing the metallic and ionic radii;[96] this effect is already seen for francium.[33]:1729–1730 This assumes that ununennium will behave chemically as an alkali metal, which, although likely, may not be true due to relativistic effects.[97] The relativistic stabilisation of the 8s orbital also increases ununennium's electron affinity far beyond that of caesium and francium; indeed, ununennium is expected to have an electron affinity higher than all the alkali metals lighter than it. Relativistic effects also cause a very large drop in the polarisability of ununennium.[33]:1729–1730 On the other hand, ununennium is predicted to continue the trend of melting points decreasing going down the group, being expected to have a melting point between 0 °C and 30 °C.[33]:1724
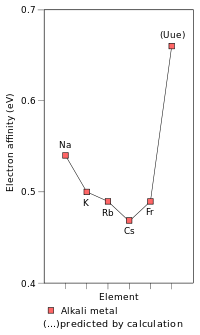
The stabilisation of ununennium's valence electron and thus the contraction of the 8s orbital cause its atomic radius to be lowered to 240 pm,[33]:1729–1730 very close to that of rubidium (247 pm),[5] so that the chemistry of ununennium in the +1 oxidation state should be more similar to the chemistry of rubidium than to that of francium. On the other hand, the ionic radius of the Uue+ ion is predicted to be larger than that of Rb+, because the 7p orbitals are destabilised and are thus larger than the p-orbitals of the lower shells. Ununennium may also show the +3 oxidation state,[33]:1729–1730 which is not seen in any other alkali metal,[6]:28 in addition to the +1 oxidation state that is characteristic of the other alkali metals and is also the main oxidation state of all the known alkali metals: this is because of the destabilisation and expansion of the 7p3/2 spinor, causing its outermost electrons to have a lower ionisation energy than what would otherwise be expected.[6]:28[33]:1729–1730 Indeed, many ununennium compounds are expected to have a large covalent character, due to the involvement of the 7p3/2 electrons in the bonding.[34]

Not as much work has been done predicting the properties of the alkali metals beyond ununennium. Although a simple extrapolation of the periodic table would put element 169, unhexennium, under ununennium, Dirac-Fock calculations predict that the next alkali metal after ununennium may actually be element 165, unhexpentium, which is predicted to have the electron configuration [Og] 5 g18 6f14 7d10 8s2 8p1/22 9s1.[33]:1729–1730[95] Furthermore, this element would be intermediate in properties between an alkali metal and a group 11 element, and while its physical and atomic properties would be closer to the former, its chemistry may be closer to that of the latter. Further calculations show that unhexpentium would follow the trend of increasing ionisation energy beyond caesium, having an ionisation energy comparable to that of sodium, and that it should also continue the trend of decreasing atomic radii beyond caesium, having an atomic radius comparable to that of potassium.[33]:1729–1730 However, the 7d electrons of unhexpentium may also be able to participate in chemical reactions along with the 9s electron, possibly allowing oxidation states beyond +1, whence the likely transition metal behaviour of unhexpentium.[33]:1732–1733[98] Due to the alkali and alkaline earth metals both being s-block elements, these predictions for the trends and properties of ununennium and unhexpentium also mostly hold quite similarly for the corresponding alkaline earth metals unbinilium (Ubn) and unhexhexium (Uhh).[33]:1729–1733
The probable properties of further alkali metals beyond unhexpentium have not been explored yet as of 2015; in fact, it is suspected that they may not be able to exist.[95] In periods 8 and above of the periodic table, relativistic and shell-structure effects become so strong that extrapolations from lighter congeners become completely inaccurate. In addition, the relativistic and shell-structure effects (which stabilise the s-orbitals and destabilise and expand the d-, f-, and g-orbitals of higher shells) have opposite effects, causing even larger difference between relativistic and non-relativistic calculations of the properties of elements with such high atomic numbers.[33]:1732–1733 Interest in the chemical properties of ununennium and unhexpentium stems from the fact that both elements are located close to the expected locations of islands of stabilities, centered at elements 122 (306Ubb) and 164 (482Uhq).[99][100][101]
Pseudo-alkali metals
Many other substances are similar to the alkali metals in their tendency to form monopositive cations. Analogously to the pseudohalogens, they have sometimes been called "pseudo-alkali metals". These substances include some elements and many more polyatomic ions; the polyatomic ions are especially similar to the alkali metals in their large size and weak polarising power.[102]
Hydrogen
The element hydrogen, with one electron per neutral atom, is usually placed at the top of Group 1 of the periodic table for convenience, but hydrogen is not normally considered to be an alkali metal;[103] when it is considered to be an alkali metal, it is because of its atomic properties and not its chemical properties.[104] Under typical conditions, pure hydrogen exists as a diatomic gas consisting of two atoms per molecule (H2);[105] however, the alkali metals only form diatomic molecules (such as dilithium, Li2) at high temperatures, when they are in the gaseous state.[106]
Hydrogen, like the alkali metals, has one valence electron[73] and reacts easily with the halogens,[73] but the similarities end there because of the small size of a bare proton H+ compared to the alkali metal cations.[73] Its placement above lithium is primarily due to its electron configuration.[103][107] It is sometimes placed above carbon due to their similar electronegativities[108] or fluorine due to their similar chemical properties.[107][108]
The first ionisation energy of hydrogen (1312.0 kJ/mol) is much higher than that of the alkali metals.[109][110] As only one additional electron is required to fill in the outermost shell of the hydrogen atom, hydrogen often behaves like a halogen, forming the negative hydride ion, and is very occasionally considered to be a halogen on that basis.[107] (The alkali metals can also form negative ions, known as alkalides, but these are little more than laboratory curiosities, being unstable.)[26][27] An argument against this placement is that formation of hydride from hydrogen is endothermic, unlike the exothermic formation of halides from halogens. The radius of the H− anion also does not fit the trend of increasing size going down the halogens: indeed, H− is very diffuse because its single proton cannot easily control both electrons.[73]:15–6 It was expected for some time that liquid hydrogen would show metallic properties;[108] while this has been shown to not be the case, under extremely high pressures, such as those found at the cores of Jupiter and Saturn, hydrogen does become metallic and behaves like an alkali metal; in this phase, it is known as metallic hydrogen.[111] The electrical resistivity of liquid metallic hydrogen at 3000 K is approximately equal to that of liquid rubidium and caesium at 2000 K at the respective pressures when they undergo a nonmetal-to-metal transition.[112]
The 1s1 electron configuration of hydrogen, while superficially similar to that of the alkali metals (ns1), is unique because there is no 1p subshell. Hence it can lose an electron to form the hydron H+, or gain one to form the hydride ion H−.[6]:43 In the former case it resembles superficially the alkali metals; in the latter case, the halogens, but the differences due to the lack of a 1p subshell are important enough that neither group fits the properties of hydrogen well.[6]:43 Group 14 is also a good fit in terms of thermodynamic properties such as ionisation energy and electron affinity, but makes chemical nonsense because hydrogen cannot be tetravalent. Thus none of the three placements are entirely satisfactory, although group 1 is the most common placement (if one is chosen) because the hydron is by far the most important of all monatomic hydrogen species, being the foundation of acid-base chemistry.[108] As an example of hydrogen's unorthodox properties stemming from its unusual electron configuration and small size, the hydrogen ion is very small (radius around 150 fm compared to the 50–220 pm size of most other atoms and ions) and so is nonexistent in condensed systems other than in association with other atoms or molecules. Indeed, transferring of protons between chemicals is the basis of acid-base chemistry.[6]:43 Also unique is hydrogen's ability to form hydrogen bonds, which are an effect of charge-transfer, electrostatic, and electron correlative contributing phenomena.[108] While analogous lithium bonds are also known, they are mostly electrostatic.[108] Nevertheless, hydrogen can take on the same structural role as the alkali metals in some molecular crystals, and has a close relationship with the lightest alkali metals (especially lithium).[113]
Ammonium and derivatives

The ammonium ion (NH+
4) has very similar properties to the heavier alkali metals, acting as an alkali metal intermediate between potassium and rubidium,[102][114] and is often considered a close relative.[115][116][117] For example, most alkali metal salts are soluble in water, a property which ammonium salts share.[118] Ammonium is expected to behave stably as a metal (NH+
4 ions in a sea of delocalised electrons) at very high pressures (though less than the typical pressure where transitions from insulating to metallic behaviour occur around, 100 GPa), and could possibly occur inside the ice giants Uranus and Neptune, which may have significant impacts on their interior magnetic fields.[116][117] It has been estimated that the transition from a mixture of ammonia and dihydrogen molecules to metallic ammonium may occur at pressures just below 25 GPa.[116] Under standard conditions, ammonium can form a metallic amalgam with mercury.[119]
Other "pseudo-alkali metals" include the alkylammonium cations, in which some of the hydrogen atoms in the ammonium cation are replaced by alkyl or aryl groups. In particular, the quaternary ammonium cations (NR+
4) are very useful since they are permanently charged, and they are often used as an alternative to the expensive Cs+ to stabilise very large and very easily polarisable anions such as HI−
2.[6]:812–9 Tetraalkylammonium hydroxides, like alkali metal hydroxides, are very strong bases that react with atmospheric carbon dioxide to form carbonates.[73]:256 Furthermore, the nitrogen atom may be replaced by a phosphorus, arsenic, or antimony atom (the heavier nonmetallic pnictogens), creating a phosphonium (PH+
4) or arsonium (AsH+
4) cation that can itself be substituted similarly; while stibonium (SbH+
4) itself is not known, some of its organic derivatives are characterised.[102]
Cobaltocene and derivatives
Cobaltocene, Co(C5H5)2, is a metallocene, the cobalt analogue of ferrocene. It is a dark purple solid. Cobaltocene has 19 valence electrons, one more than usually found in organotransition metal complexes, such as its very stable relative, ferrocene, in accordance with the 18-electron rule. This additional electron occupies an orbital that is antibonding with respect to the Co–C bonds. Consequently, many chemical reactions of Co(C5H5)2 are characterized by its tendency to lose this "extra" electron, yielding a very stable 18-electron cation known as cobaltocenium. Many cobaltocenium salts coprecipitate with caesium salts, and cobaltocenium hydroxide is a strong base that absorbs atmospheric carbon dioxide to form cobaltocenium carbonate.[73]:256 Like the alkali metals, cobaltocene is a strong reducing agent, and decamethylcobaltocene is stronger still due to the combined inductive effect of the ten methyl groups.[120] Cobalt may be substituted by its heavier congener rhodium to give rhodocene, an even stronger reducing agent.[121] Iridocene (involving iridium) would presumably be still more potent, but is not very well-studied due to its instability.[122]
Thallium

Thallium is the heaviest stable element in group 13 of the periodic table. At the bottom of the periodic table, the inert pair effect is quite strong, because of the relativistic stabilisation of the 6s orbital and the decreasing bond energy as the atoms increase in size so that the amount of energy released in forming two more bonds is not worth the high ionisation energies of the 6s electrons.[6]:226–7 It displays the +1 oxidation state[6]:28 that all the known alkali metals display,[6]:28 and thallium compounds with thallium in its +1 oxidation state closely resemble the corresponding potassium or silver compounds stoichiometrically due to the similar ionic radii of the Tl+ (164 pm), K+ (152 pm) and Ag+ (129 pm) ions.[123][124] It was sometimes considered an alkali metal in continental Europe (but not in England) in the years immediately following its discovery,[124]:126 and was placed just after caesium as the sixth alkali metal in Dmitri Mendeleev's 1869 periodic table and Julius Lothar Meyer's 1868 periodic table.[125] (Mendeleev's 1871 periodic table and Meyer's 1870 periodic table put thallium in its current position in the boron group and left the space below caesium blank.)[125] However, thallium also displays the oxidation state +3,[6]:28 which no known alkali metal displays[6]:28 (although ununennium, the undiscovered seventh alkali metal, is predicted to possibly display the +3 oxidation state).[33]:1729–1730 The sixth alkali metal is now considered to be francium.[126] While Tl+ is stabilised by the inert pair effect, this inert pair of 6s electrons is still able to participate chemically, so that these electrons are stereochemically active in aqueous solution. Additionally, the thallium halides (except TlF) are quite insoluble in water, and TlI has an unusual structure because of the presence of the stereochemically active inert pair in thallium.[127]
Copper, silver, and gold



The group 11 metals (or coinage metals), copper, silver, and gold, are typically categorised as transition metals given they can form ions with incomplete d-shells. Physically, they have the relatively low melting points and high electronegativity values associated with post-transition metals. "The filled d subshell and free s electron of Cu, Ag, and Au contribute to their high electrical and thermal conductivity. Transition metals to the left of group 11 experience interactions between s electrons and the partially filled d subshell that lower electron mobility."[128] Chemically, the group 11 metals behave like main-group metals in their +1 valence states, and are hence somewhat related to the alkali metals: this is one reason for their previously being labelled as "group IB", paralleling the alkali metals' "group IA". They are occasionally classified as post-transition metals.[129] Their spectra are analogous to those of the alkali metals.[130] Their monopositive ions are paramagnetic and contribute no colour to their salts, like those of the alkali metals.[131]
In Mendeleev's 1871 periodic table, copper, silver, and gold are listed twice, once under group VIII (with the iron triad and platinum group metals), and once under group IB. Group IB was nonetheless parenthesised to note that it was tentative. Mendeleev's main criterion for group assignment was the maximum oxidation state of an element: on that basis, the group 11 elements could not be classified in group IB, due to the existence of copper(II) and gold(III) compounds being known at that time.[130] However, eliminating group IB would make group I the only main group (group VIII was labelled a transition group) to lack an A–B bifurcation.[130] Soon afterward, a majority of chemists chose to classify these elements in group IB and remove them from group VIII for the resulting symmetry: this was the predominant classification until the rise of the modern medium-long 18-column periodic table, which separated the alkali metals and group 11 metals.[130]
The coinage metals were traditionally regarded as a subdivision of the alkali metal group, due to them sharing the characteristic s1 electron configuration of the alkali metals (group 1: p6s1; group 11: d10s1). However, the similarities are largely confined to the stoichiometries of the +1 compounds of both groups, and not their chemical properties.[6]:1177 This stems from the filled d subshell providing a much weaker shielding effect on the outermost s electron than the filled p subshell, so that the coinage metals have much higher first ionisation energies and smaller ionic radii than do the corresponding alkali metals.[6]:1177 Furthermore, they have higher melting points, hardnesses, and densities, and lower reactivities and solubilities in liquid ammonia, as well as having more covalent character in their compounds.[6]:1177 Finally, the alkali metals are at the top of the electrochemical series, whereas the coinage metals are almost at the very bottom.[6]:1177 The coinage metals' filled d shell is much more easily disrupted than the alkali metals' filled p shell, so that the second and third ionisation energies are lower, enabling higher oxidation states than +1 and a richer coordination chemistry, thus giving the group 11 metals clear transition metal character.[6]:1177 Particularly noteworthy is gold forming ionic compounds with rubidium and caesium, in which it forms the auride ion (Au−) which also occurs in solvated form in liquid ammonia solution: here gold behaves as a pseudohalogen because its 5d106s1 configuration has one electron less than the quasi-closed shell 5d106s2 configuration of mercury.[6]:1177
History

Sodium compounds have been known since ancient times; salt (sodium chloride) has been an important commodity in human activities, as testified by the English word salary, referring to salarium, money paid to Roman soldiers for the purchase of salt.[132] While potash has been used since ancient times, it was not understood for most of its history to be a fundamentally different substance from sodium mineral salts. Georg Ernst Stahl obtained experimental evidence which led him to suggest the fundamental difference of sodium and potassium salts in 1702,[133] and Henri Louis Duhamel du Monceau was able to prove this difference in 1736.[134] The exact chemical composition of potassium and sodium compounds, and the status as chemical element of potassium and sodium, was not known then, and thus Antoine Lavoisier did include the alkali in his list of chemical elements in 1789.[135][136]
Pure potassium was first isolated in 1807 in England by Sir Humphry Davy, who derived it from caustic potash (KOH, potassium hydroxide) by the use of electrolysis of the molten salt with the newly invented voltaic pile. Previous attempts at electrolysis of the aqueous salt were unsuccessful due to potassium's extreme reactivity.[6]:68 Potassium was the first metal that was isolated by electrolysis.[137] Later that same year, Davy reported extraction of sodium from the similar substance caustic soda (NaOH, lye) by a similar technique, demonstrating the elements, and thus the salts, to be different.[135][136][138][139] Later that year, the first pieces of pure molten sodium metal were similarly prepared by Humphry Davy through the electrolysis of molten caustic soda (now called sodium hydroxide).[138]

Petalite (LiAlSi4O10) was discovered in 1800 by the Brazilian chemist José Bonifácio de Andrada in a mine on the island of Utö, Sweden.[140][141][142] However, it was not until 1817 that Johan August Arfwedson, then working in the laboratory of the chemist Jöns Jacob Berzelius, detected the presence of a new element while analysing petalite ore.[143][144] This new element was noted by him to form compounds similar to those of sodium and potassium, though its carbonate and hydroxide were less soluble in water and more alkaline than the other alkali metals.[145] Berzelius gave the unknown material the name "lithion/lithina", from the Greek word λιθoς (transliterated as lithos, meaning "stone"), to reflect its discovery in a solid mineral, as opposed to potassium, which had been discovered in plant ashes, and sodium, which was known partly for its high abundance in animal blood. He named the metal inside the material "lithium".[18][141][144] Lithium, sodium, and potassium were part of the discovery of periodicity, as they are among a series of triads of elements in the same group that were noted by Johann Wolfgang Döbereiner in 1850 as having similar properties.[125]

Rubidium and caesium were the first elements to be discovered using the spectroscope, invented in 1859 by Robert Bunsen and Gustav Kirchhoff.[146] The next year, they discovered caesium in the mineral water from Bad Dürkheim, Germany. Their discovery of rubidium came the following year in Heidelberg, Germany, finding it in the mineral lepidolite.[147] The names of rubidium and caesium come from the most prominent lines in their emission spectra: a bright red line for rubidium (from the Latin word rubidus, meaning dark red or bright red), and a sky-blue line for caesium (derived from the Latin word caesius, meaning sky-blue).[148][149]
Around 1865 John Newlands produced a series of papers where he listed the elements in order of increasing atomic weight and similar physical and chemical properties that recurred at intervals of eight; he likened such periodicity to the octaves of music, where notes an octave apart have similar musical functions.[150][151] His version put all the alkali metals then known (lithium to caesium), as well as copper, silver, and thallium (which show the +1 oxidation state characteristic of the alkali metals), together into a group. His table placed hydrogen with the halogens.[125]

After 1869, Dmitri Mendeleev proposed his periodic table placing lithium at the top of a group with sodium, potassium, rubidium, caesium, and thallium.[152] Two years later, Mendeleev revised his table, placing hydrogen in group 1 above lithium, and also moving thallium to the boron group. In this 1871 version, copper, silver, and gold were placed twice, once as part of group IB, and once as part of a "group VIII" encompassing today's groups 8 to 11.[130][note 8] After the introduction of the 18-column table, the group IB elements were moved to their current position in the d-block, while alkali metals were left in group IA. Later the group's name was changed to group 1 in 1988.[153] The trivial name "alkali metals" comes from the fact that the hydroxides of the group 1 elements are all strong alkalis when dissolved in water.[5]
There were at least four erroneous and incomplete discoveries[43][44][154][155] before Marguerite Perey of the Curie Institute in Paris, France discovered francium in 1939 by purifying a sample of actinium-227, which had been reported to have a decay energy of 220 keV. However, Perey noticed decay particles with an energy level below 80 keV. Perey thought this decay activity might have been caused by a previously unidentified decay product, one that was separated during purification, but emerged again out of the pure actinium-227. Various tests eliminated the possibility of the unknown element being thorium, radium, lead, bismuth, or thallium. The new product exhibited chemical properties of an alkali metal (such as coprecipitating with caesium salts), which led Perey to believe that it was element 87, caused by the alpha decay of actinium-227.[156] Perey then attempted to determine the proportion of beta decay to alpha decay in actinium-227. Her first test put the alpha branching at 0.6%, a figure that she later revised to 1%.[157]
- 227
89Ac
223
87Fr
223
88Ra
The next element below francium (eka-francium) in the periodic table would be ununennium (Uue), element 119.[33]:1729–1730 The synthesis of ununennium was first attempted in 1985 by bombarding a target of einsteinium-254 with calcium-48 ions at the superHILAC accelerator at Berkeley, California. No atoms were identified, leading to a limiting yield of 300 nb.[158][159]
It is highly unlikely[158] that this reaction will be able to create any atoms of ununennium in the near future, given the extremely difficult task of making sufficient amounts of einsteinium-254, which is favoured for production of ultraheavy elements because of its large mass, relatively long half-life of 270 days, and availability in significant amounts of several micrograms,[160] to make a large enough target to increase the sensitivity of the experiment to the required level; einsteinium has not been found in nature and has only been produced in laboratories, and in quantities smaller than those needed for effective synthesis of ultraheavy elements. However, given that ununennium is only the first period 8 element on the extended periodic table, it may well be discovered in the near future through other reactions, and indeed attempts to synthesise it in 2019 and 2020 are currently planned at laboratories at Japan and Russia.[161] Currently, none of the period 8 elements have been discovered yet, and it is also possible, due to drip instabilities, that only the lower period 8 elements, up to around element 128, are physically possible.[96][162] No attempts at synthesis have been made for any heavier alkali metals, such as unhexpentium, due to their extremely high atomic number: they would require new, more powerful technology to make.[33]:1737–1739
Occurrence
In the Solar System

The Oddo–Harkins rule holds that elements with even atomic numbers are more common that those with odd atomic numbers, with the exception of hydrogen. This rule argues that elements with odd atomic numbers have one unpaired proton and are more likely to capture another, thus increasing their atomic number. In elements with even atomic numbers, protons are paired, with each member of the pair offsetting the spin of the other, enhancing stability.[164][165][166] All the alkali metals have odd atomic numbers and they are not as common as the elements with even atomic numbers adjacent to them (the noble gases and the alkaline earth metals) in the Solar System. The heavier alkali metals are also less abundant than the lighter ones as the alkali metals from rubidium onward can only be synthesised in supernovae and not in stellar nucleosynthesis. Lithium is also much less abundant than sodium and potassium as it is poorly synthesised in both Big Bang nucleosynthesis and in stars: the Big Bang could only produce trace quantities of lithium, beryllium and boron due to the absence of a stable nucleus with 5 or 8 nucleons, and stellar nucleosynthesis could only pass this bottleneck by the triple-alpha process, fusing three helium nuclei to form carbon, and skipping over those three elements.[163]
On Earth

The Earth formed from the same cloud of matter that formed the Sun, but the planets acquired different compositions during the formation and evolution of the solar system. In turn, the natural history of the Earth caused parts of this planet to have differing concentrations of the elements. The mass of the Earth is approximately 5.98×1024 kg. It is composed mostly of iron (32.1%), oxygen (30.1%), silicon (15.1%), magnesium (13.9%), sulfur (2.9%), nickel (1.8%), calcium (1.5%), and aluminium (1.4%); with the remaining 1.2% consisting of trace amounts of other elements. Due to planetary differentiation, the core region is believed to be primarily composed of iron (88.8%), with smaller amounts of nickel (5.8%), sulfur (4.5%), and less than 1% trace elements.[167]
The alkali metals, due to their high reactivity, do not occur naturally in pure form in nature. They are lithophiles and therefore remain close to the Earth's surface because they combine readily with oxygen and so associate strongly with silica, forming relatively low-density minerals that do not sink down into the Earth's core. Potassium, rubidium and caesium are also incompatible elements due to their large ionic radii.[168]
Sodium and potassium are very abundant in earth, both being among the ten most common elements in Earth's crust;[169][170] sodium makes up approximately 2.6% of the Earth's crust measured by weight, making it the sixth most abundant element overall[171] and the most abundant alkali metal. Potassium makes up approximately 1.5% of the Earth's crust and is the seventh most abundant element.[171] Sodium is found in many different minerals, of which the most common is ordinary salt (sodium chloride), which occurs in vast quantities dissolved in seawater. Other solid deposits include halite, amphibole, cryolite, nitratine, and zeolite.[171] Many of these solid deposits occur as a result of ancient seas evaporating, which still occurs now in places such as Utah's Great Salt Lake and the Dead Sea.[6]:69 Despite their near-equal abundance in Earth's crust, sodium is far more common than potassium in the ocean, both because potassium's larger size makes its salts less soluble, and because potassium is bound by silicates in soil and what potassium leaches is absorbed far more readily by plant life than sodium.[6]:69
Despite its chemical similarity, lithium typically does not occur together with sodium or potassium due to its smaller size.[6]:69 Due to its relatively low reactivity, it can be found in seawater in large amounts; it is estimated that seawater is approximately 0.14 to 0.25 parts per million (ppm)[172][173] or 25 micromolar.[174] Its diagonal relationship with magnesium often allows it to replace magnesium in ferromagnesium minerals, where its crustal concentration is about 18 ppm, comparable to that of gallium and niobium. Commercially, the most important lithium mineral is spodumene, which occurs in large deposits worldwide.[6]:69
Rubidium is approximately as abundant as zinc and more abundant than copper. It occurs naturally in the minerals leucite, pollucite, carnallite, zinnwaldite, and lepidolite,[175] although none of these contain only rubidium and no other alkali metals.[6]:70 Caesium is more abundant than some commonly known elements, such as antimony, cadmium, tin, and tungsten, but is much less abundant than rubidium.[21]
Francium-223, the only naturally occurring isotope of francium,[10][11] is the product of the alpha decay of actinium-227 and can be found in trace amounts in uranium minerals.[176] In a given sample of uranium, there is estimated to be only one francium atom for every 1018 uranium atoms.[177][178] It has been calculated that there is at most 30 g of francium in the earth's crust at any time, due to its extremely short half-life of 22 minutes.[179][180]
Production and isolation


The production of pure alkali metals is somewhat complicated due to their extreme reactivity with commonly used substances, such as water.[5][8] From their silicate ores, all the stable alkali metals may be obtained the same way: sulfuric acid is first used to dissolve the desired alkali metal ion and aluminium(III) ions from the ore (leaching), whereupon basic precipitation removes aluminium ions from the mixture by precipitating it as the hydroxide. The remaining insoluble alkali metal carbonate is then precipitated selectively; the salt is then dissolved in hydrochloric acid to produce the chloride. The result is then left to evaporate and the alkali metal can then be isolated.[8] Lithium and sodium are typically isolated through electrolysis from their liquid chlorides, with calcium chloride typically added to lower the melting point of the mixture. The heavier alkali metals, however, is more typically isolated in a different way, where a reducing agent (typically sodium for potassium and magnesium or calcium for the heaviest alkali metals) is used to reduce the alkali metal chloride. The liquid or gaseous product (the alkali metal) then undergoes fractional distillation for purification.[8]
Lithium salts have to be extracted from the water of mineral springs, brine pools, and brine deposits. The metal is produced electrolytically from a mixture of fused lithium chloride and potassium chloride.[181]
Sodium occurs mostly in seawater and dried seabed,[5] but is now produced through electrolysis of sodium chloride by lowering the melting point of the substance to below 700 °C through the use of a Downs cell.[182][183] Extremely pure sodium can be produced through the thermal decomposition of sodium azide.[184] Potassium occurs in many minerals, such as sylvite (potassium chloride).[5] Previously, potassium was generally made from the electrolysis of potassium chloride or potassium hydroxide,[185] found extensively in places such as Canada, Russia, Belarus, Germany, Israel, United States, and Jordan, in a method similar to how sodium was produced in the late 1800s and early 1900s.[186] It can also be produced from seawater.[5] However, these methods are problematic because the potassium metal tends to dissolve in its molten chloride and vaporises significantly at the operating temperatures, potentially forming the explosive superoxide. As a result, pure potassium metal is now produced by reducing molten potassium chloride with sodium metal at 850 °C.[6]:74
- Na (g) + KCl (l) ⇌ NaCl (l) + K (g)
Although sodium is less reactive than potassium, this process works because at such high temperatures potassium is more volatile than sodium and can easily be distilled off, so that the equilibrium shifts towards the right to produce more potassium gas and proceeds almost to completion.[6]:74

For several years in the 1950s and 1960s, a by-product of the potassium production called Alkarb was a main source for rubidium. Alkarb contained 21% rubidium while the rest was potassium and a small fraction of caesium.[187] Today the largest producers of caesium, for example the Tanco Mine in Manitoba, Canada, produce rubidium as by-product from pollucite.[188] Today, a common method for separating rubidium from potassium and caesium is the fractional crystallisation of a rubidium and caesium alum (Cs,Rb)Al(SO4)2·12H2O, which yields pure rubidium alum after approximately 30 recrystallisations.[188][189] The limited applications and the lack of a mineral rich in rubidium limit the production of rubidium compounds to 2 to 4 tonnes per year.[188] Caesium, however, is not produced from the above reaction. Instead, the mining of pollucite ore is the main method of obtaining pure caesium, extracted from the ore mainly by three methods: acid digestion, alkaline decomposition, and direct reduction.[188][190] Both metals are produced as by-products of lithium production: after 1958, when interest in lithium's thermonuclear properties increased sharply, the production of rubidium and caesium also increased correspondingly.[6]:71 Pure rubidium and caesium metals are produced by reducing their chlorides with calcium metal at 750 °C and low pressure.[6]:74
As a result of its extreme rarity in nature,[179] most francium is synthesised in the nuclear reaction 197Au + 18O → 210Fr + 5 n, yielding francium-209, francium-210, and francium-211.[191] The greatest quantity of francium ever assembled to date is about 300,000 neutral atoms,[192] which were synthesised using the nuclear reaction given above.[192] When the only natural isotope francium-223 is specifically required, it is produced as the alpha daughter of actinium-227, itself produced synthetically from the neutron irradiation of natural radium-226, one of the daughters of natural uranium-238.[193]
Applications

Lithium, sodium, and potassium have many applications, while rubidium and caesium are very useful in academic contexts but do not have many applications yet.[6]:68 Lithium is often used in batteries, and lithium oxide can help process silica. Lithium stearate is a thickener and can be used to make lubricating greases; it is produced from lithium hydroxide, which is also used to absorb carbon dioxide in space capsules and submarines.[6]:70 Lithium chloride is used as a brazing alloy for aluminium parts.[194] Metallic lithium is used in alloys with magnesium and aluminium to give very tough and light alloys.[6]:70
Sodium compounds have many applications, the most well-known being sodium chloride as table salt. Sodium salts of fatty acids are used as soap.[195] Pure sodium metal also has many applications, including use in sodium-vapour lamps, which produce very efficient light compared to other types of lighting,[196][197] and can help smooth the surface of other metals.[198][199] Being a strong reducing agent, it is often used to reduce many other metals, such as titanium and zirconium, from their chlorides. Furthermore, it is very useful as a heat-exchange liquid in fast breeder nuclear reactors due to its low melting point, viscosity, and cross-section towards neutron absorption.[6]:74
Potassium compounds are often used as fertilisers[6]:73[200] as potassium is an important element for plant nutrition. Potassium hydroxide is a very strong base, and is used to control the pH of various substances.[201][202] Potassium nitrate and potassium permanganate are often used as powerful oxidising agents.[6]:73 Potassium superoxide is used in breathing masks, as it reacts with carbon dioxide to give potassium carbonate and oxygen gas. Pure potassium metal is not often used, but its alloys with sodium may substitute for pure sodium in fast breeder nuclear reactors.[6]:74
Rubidium and caesium are often used in atomic clocks.[203] Caesium atomic clocks are extraordinarily accurate; if a clock had been made at the time of the dinosaurs, it would be off by less than four seconds (after 80 million years).[21] For that reason, caesium atoms are used as the definition of the second.[204] Rubidium ions are often used in purple fireworks,[205] and caesium is often used in drilling fluids in the petroleum industry.[21][206]
Francium has no commercial applications,[177][178][207] but because of francium's relatively simple atomic structure, among other things, it has been used in spectroscopy experiments, leading to more information regarding energy levels and the coupling constants between subatomic particles.[208] Studies on the light emitted by laser-trapped francium-210 ions have provided accurate data on transitions between atomic energy levels, similar to those predicted by quantum theory.[209]
Biological role and precautions
Metals
Pure alkali metals are dangerously reactive with air and water and must be kept away from heat, fire, oxidising agents, acids, most organic compounds, halocarbons, plastics, and moisture. They also react with carbon dioxide and carbon tetrachloride, so that normal fire extinguishers are counterproductive when used on alkali metal fires.[210] Some Class D dry powder extinguishers designed for metal fires are effective, depriving the fire of oxygen and cooling the alkali metal.[211]
Experiments are usually conducted using only small quantities of a few grams in a fume hood. Small quantities of lithium may be disposed of by reaction with cool water, but the heavier alkali metals should be dissolved in the less reactive isopropanol.[210][212] The alkali metals must be stored under mineral oil or an inert atmosphere. The inert atmosphere used may be argon or nitrogen gas, except for lithium, which reacts with nitrogen.[210] Rubidium and caesium must be kept away from air, even under oil, because even a small amount of air diffused into the oil may trigger formation of the dangerously explosive peroxide; for the same reason, potassium should not be stored under oil in an oxygen-containing atmosphere for longer than 6 months.[213][214]
Ions

The bioinorganic chemistry of the alkali metal ions has been extensively reviewed.[215] Solid state crystal structures have been determined for many complexes of alkali metal ions in small peptides, nucleic acid constituents, carbohydrates and ionophore complexes.[216]
Lithium naturally only occurs in traces in biological systems and has no known biological role, but does have effects on the body when ingested.[217] Lithium carbonate is used as a mood stabiliser in psychiatry to treat bipolar disorder (manic-depression) in daily doses of about 0.5 to 2 grams, although there are side-effects.[217] Excessive ingestion of lithium causes drowsiness, slurred speech and vomiting, among other symptoms,[217] and poisons the central nervous system,[217] which is dangerous as the required dosage of lithium to treat bipolar disorder is only slightly lower than the toxic dosage.[217][218] Its biochemistry, the way it is handled by the human body and studies using rats and goats suggest that it is an essential trace element, although the natural biological function of lithium in humans has yet to be identified.[219][220]
Sodium and potassium occur in all known biological systems, generally functioning as electrolytes inside and outside cells.[221][222] Sodium is an essential nutrient that regulates blood volume, blood pressure, osmotic equilibrium and pH; the minimum physiological requirement for sodium is 500 milligrams per day.[223] Sodium chloride (also known as common salt) is the principal source of sodium in the diet, and is used as seasoning and preservative, such as for pickling and jerky; most of it comes from processed foods.[224] The Dietary Reference Intake for sodium is 1.5 grams per day,[225] but most people in the United States consume more than 2.3 grams per day,[226] the minimum amount that promotes hypertension;[227] this in turn causes 7.6 million premature deaths worldwide.[228]
Potassium is the major cation (positive ion) inside animal cells,[221] while sodium is the major cation outside animal cells.[221][222] The concentration differences of these charged particles causes a difference in electric potential between the inside and outside of cells, known as the membrane potential. The balance between potassium and sodium is maintained by ion transporter proteins in the cell membrane.[229] The cell membrane potential created by potassium and sodium ions allows the cell to generate an action potential—a "spike" of electrical discharge. The ability of cells to produce electrical discharge is critical for body functions such as neurotransmission, muscle contraction, and heart function.[229] Disruption of this balance may thus be fatal: for example, ingestion of large amounts of potassium compounds can lead to hyperkalemia strongly influencing the cardiovascular system.[230][231] Potassium chloride is used in the United States for lethal injection executions.[230]

Due to their similar atomic radii, rubidium and caesium in the body mimic potassium and are taken up similarly. Rubidium has no known biological role, but may help stimulate metabolism,[232][233][234] and, similarly to caesium,[232][235] replace potassium in the body causing potassium deficiency.[232][234] Partial substitution is quite possible and rather non-toxic: a 70 kg person contains on average 0.36 g of rubidium, and an increase in this value by 50 to 100 times did not show negative effects in test persons.[236] Rats can survive up to 50% substitution of potassium by rubidium.[237][238] Rubidium (and to a much lesser extent caesium) can function as temporary cures for hypokalemia; while rubidium can adequately physiologically substitute potassium in some systems, caesium is never able to do so.[233] There is only very limited evidence in the form of deficiency symptoms for rubidium being possibly essential in goats; even if this is true, the trace amounts usually present in food are more than enough.[239][240]
Caesium compounds are rarely encountered by most people, but most caesium compounds are mildly toxic. Like rubidium, caesium tends to substitute potassium in the body, but is significantly larger and is therefore a poorer substitute.[235] Excess caesium can lead to hypokalemia, arrythmia, and acute cardiac arrest,[241] but such amounts would not ordinarily be encountered in natural sources.[242] As such, caesium is not a major chemical environmental pollutant.[243] The median lethal dose (LD50) value for caesium chloride in mice is 2.3 g per kilogram, which is comparable to the LD50 values of potassium chloride and sodium chloride.[244] Caesium chloride has been promoted as an alternative cancer therapy,[245] but has been linked to the deaths of over 50 patients, on whom it was used as part of a scientifically unvalidated cancer treatment.[246]
Radioisotopes of caesium require special precautions: the improper handling of caesium-137 gamma ray sources can lead to release of this radioisotope and radiation injuries. Perhaps the best-known case is the Goiânia accident of 1987, in which an improperly-disposed-of radiation therapy system from an abandoned clinic in the city of Goiânia, Brazil, was scavenged from a junkyard, and the glowing caesium salt sold to curious, uneducated buyers. This led to four deaths and serious injuries from radiation exposure. Together with caesium-134, iodine-131, and strontium-90, caesium-137 was among the isotopes distributed by the Chernobyl disaster which constitute the greatest risk to health.[47] Radioisotopes of francium would presumably be dangerous as well due to their high decay energy and short half-life, but none have been produced in large enough amounts to pose any serious risk.[193]
Notes
- ↑ The symbols Na and K for sodium and potassium are derived from their Latin names, natrium and kalium; these are still the names for the elements in some languages, such as German and Russian.
- ↑ Caesium is the spelling recommended by the International Union of Pure and Applied Chemistry (IUPAC).[1] The American Chemical Society (ACS) has used the spelling cesium since 1921,[2][3] following Webster’s Third New International Dictionary.
- ↑ In both the old IUPAC and the CAS systems for group numbering, this group is known as group IA (pronounced as "group one A", as the "I" is a Roman numeral).[4]
- ↑ The number given in parentheses refers to the measurement uncertainty. This uncertainty applies to the least significant figure(s) of the number prior to the parenthesised value (ie. counting from rightmost digit to left). For instance, 1.00794(7) stands for 1.00794±0.00007, while 1.00794(72) stands for 1.00794±0.00072.[9]
- ↑ The value listed is the conventional value suitable for trade and commerce; the actual value may range from 6.938 to 6.997 depending on the isotopic composition of the sample.[11]
- ↑ The element does not have any stable nuclides, and a value in brackets indicates the mass number of the longest-lived isotope of the element.[10][11]
- ↑ Linus Pauling estimated the electronegativity of francium at 0.7 on the Pauling scale, the same as caesium;[13] the value for caesium has since been refined to 0.79, although there are no experimental data to allow a refinement of the value for francium.[14] Francium has a slightly higher ionisation energy than caesium,[12] 392.811(4) kJ/mol as opposed to 375.7041(2) kJ/mol for caesium, as would be expected from relativistic effects, and this would imply that caesium is the less electronegative of the two.
- ↑ In the 1869 version of Mendeleev's periodic table, copper and silver were placed in their own group, aligned with hydrogen and mercury, while gold was tentatively placed under uranium and the undiscovered eka-aluminium in the boron group.
- ↑ The asterisk denotes an excited state.
References
- ↑ International Union of Pure and Applied Chemistry (2005). Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005). Cambridge (UK): RSC–IUPAC. ISBN 0-85404-438-8. pp. 248–49. Electronic version..
- ↑ Coghill, Anne M.; Garson, Lorrin R., eds. (2006). The ACS Style Guide: Effective Communication of Scientific Information (3rd ed.). Washington, D.C.: American Chemical Society. p. 127. ISBN 0-8412-3999-1.
- ↑ Coplen, T. B.; Peiser, H. S. (1998). "History of the recommended atomic-weight values from 1882 to 1997: a comparison of differences from current values to the estimated uncertainties of earlier values" (PDF). Pure Appl. Chem. 70 (1): 237–257. doi:10.1351/pac199870010237.
- ↑ Fluck, E. (1988). "New Notations in the Periodic Table" (PDF). Pure Appl. Chem. IUPAC. 60 (3): 431–436. doi:10.1351/pac198860030431. Retrieved 24 March 2012.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 Royal Society of Chemistry. "Visual Elements: Group 1 – The Alkali Metals". Visual Elements. Royal Society of Chemistry. Archived from the original on 5 August 2012. Retrieved 13 January 2012.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 0-08-037941-9.
- 1 2 3 Lide, D. R., ed. (2003). CRC Handbook of Chemistry and Physics (84th ed.). Boca Raton, FL: CRC Press.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 Averill, Bruce A.; Eldredge, Patricia (2007). "21.3: The Alkali Metals". Chemistry: Principles, Patterns, and Applications with Student Access Kit for Mastering General Chemistry (1st ed.). Prentice Hall. ISBN 978-0-8053-3799-0. Retrieved 24 June 2013.
- ↑ "Standard Uncertainty and Relative Standard Uncertainty". CODATA reference. National Institute of Standards and Technology. Retrieved 26 September 2011.
- 1 2 3 Wieser, Michael E.; Berglund, Michael (2009). "Atomic weights of the elements 2007 (IUPAC Technical Report)" (PDF). Pure Appl. Chem. IUPAC. 81 (11): 2131–2156. doi:10.1351/PAC-REP-09-08-03. Retrieved 7 February 2012.
- 1 2 3 4 Wieser, Michael E.; Coplen, Tyler B. (2011). "Atomic weights of the elements 2009 (IUPAC Technical Report)" (PDF). Pure Appl. Chem. IUPAC. 83 (2): 359–396. doi:10.1351/PAC-REP-10-09-14. Retrieved 11 February 2012.
- 1 2 3 Andreev, S.V.; Letokhov, V.S.; Mishin, V.I. (1987). "Laser resonance photoionization spectroscopy of Rydberg levels in Fr". Phys. Rev. Lett. 59 (12): 1274–76. Bibcode:1987PhRvL..59.1274A. PMID 10035190. doi:10.1103/PhysRevLett.59.1274.
- ↑ Pauling, Linus (1960). The Nature of the Chemical Bond (Third ed.). Cornell University Press. p. 93. ISBN 978-0-8014-0333-0.
- ↑ Allred, A. L. (1961). "Electronegativity values from thermochemical data". J. Inorg. Nucl. Chem. 17 (3–4): 215–221. doi:10.1016/0022-1902(61)80142-5.
- ↑ Vanýsek, Petr (2011). “Electrochemical Series”, in Handbook of Chemistry and Physics: 92nd Edition (Chemical Rubber Company).
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 Clark, Jim (2005). "Atomic and Physical Properties of the Group 1 Elements". chemguide. Retrieved 30 January 2012.
- ↑ Gray, Theodore. "Facts, pictures, stories about the element Cesium in the Periodic Table". The Wooden Periodic Table Table. Retrieved 13 January 2012.
- 1 2 Krebs, Robert E. (2006). The History and Use of Our Earth's Chemical Elements: A Reference Guide. Westport, Conn.: Greenwood Press. ISBN 0-313-33438-2.
- ↑ The OpenLearn team (2012). "Alkali metals". OpenLearn. The Open University. Retrieved 9 July 2012.
- 1 2 Gray, Theodore. "Alkali Metal Bangs". Theodore Gray. Retrieved 13 May 2012.
- 1 2 3 4 Butterman, William C.; Brooks, William E.; Reese, Jr., Robert G. (2004). "Mineral Commodity Profile: Cesium" (PDF). United States Geological Survey. Archived from the original (PDF) on 22 November 2009. Retrieved 27 December 2009.
- ↑ Dye, James L.; Ceraso, Joseph M.; Lok, Mei; Barnett, B. L.; Tehan, Frederick J. (1974). "Crystalline salt of the sodium anion (Na−)". J. Am. Chem. Soc. 96 (2): 608–609. doi:10.1021/ja00809a060.
- ↑ Tehan, Frederick J.; Barnett, B. L.; Dye, James L. (1974). "Alkali anions. Preparation and crystal structure of a compound which contains the cryptated sodium cation and the sodium anion". J. Am. Chem. Soc. 96 (23): 7203–7208. doi:10.1021/ja00830a005.
- ↑ Dye, J. L. (1979). "Compounds of Alkali Metal Anions". Angew. Chem. Int. Ed. Engl. 18 (8): 587–598. doi:10.1002/anie.197905871.
- ↑ Redko, M. Y.; Huang, R. H.; Jackson, J. E.; Harrison, J. F.; Dye, J. L. (2003). "Barium azacryptand sodide, the first alkalide with an alkaline Earth cation, also contains a novel dimer, (Na2)2−". J. Am. Chem. Soc. 125 (8): 2259–2263. PMID 12590555. doi:10.1021/ja027241m.
- 1 2 3 ""Inverse sodium hydride": a crystalline salt that contains H+ and Na−". J. Am. Chem. Soc. 124 (21): 5928–5929. 2002. doi:10.1021/ja025655.
- 1 2 Sawicka, A.; Skurski, P.; Simons, J. (2003). "Inverse Sodium Hydride: A Theoretical Study" (PDF). J. Am. Chem. Soc. 125 (13): 3954–3958. PMID 12656631. doi:10.1021/ja021136v.
- ↑ Burgess, John (1978). Metal Ions in Solution. Chichester: Ellis Horwood. p. 20. ISBN 0-85312-027-7.
- 1 2 Richens, David. T. (1997). The Chemistry of Aqua Ions. Wiley. ISBN 0-471-97058-1.
- ↑ Persson, Ingmar (2010). "Hydrated metal ions in aqueous solution: How regular are their structures?" (PDF). Pure Appl. Chem. 82 (10): 1901–1917. doi:10.1351/PAC-CON-09-10-22. Retrieved 23 August 2014.
- 1 2 3 4 5 Clark, Jim (2005). "Reaction of the Group 1 Elements with Oxygen and Chlorine". chemguide. Retrieved 27 June 2012.
- ↑ Shriver, Duward; Atkins, Peter (2006). Inorganic Chemistry. W. H. Freeman. p. 259. ISBN 978-0-7167-4878-6. Retrieved 10 November 2012.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 Hoffman, Darleane C.; Lee, Diana M.; Pershina, Valeria (2006). "Transactinides and the future elements". In Morss; Edelstein, Norman M.; Fuger, Jean. The Chemistry of the Actinide and Transactinide Elements (3rd ed.). Dordrecht, The Netherlands: Springer. ISBN 1-4020-3555-1.
- 1 2 3 4 5 Thayer, John S. (2010). "Relativistic Effects and the Chemistry of the Heavier Main Group Elements". Relativistic Methods for Chemists: 81, 84. doi:10.1007/978-1-4020-9975-5_2.
- ↑ Landau, A.; Eliav, E.; Ishikawa, Y.; Kaldor, U. (2001). "Benchmark calculations of electron affinities of the alkali atoms sodium to eka-francium (element 119)". J. Chem. Phys. 115: 2389. doi:10.1063/1.1386413.
- ↑ Jones, Cameron; Mountford, Philip; Stasch, Andreas; Blake, Matthew P. (22 June 2015). "s-block Metal-Metal Bonds". In Liddle, Stephen T. Molecular Metal-Metal Bonds: Compounds, Synthesis, Properties. John Wiley and Sons. pp. 23–24. ISBN 978-3-527-33541-1.
- 1 2 Various authors (2002). Lide, David R., ed. Handbook of Chemistry & Physics (88th ed.). CRC. ISBN 0-8493-0486-5. OCLC 179976746. Retrieved 2008-05-23.
- ↑ "Universal Nuclide Chart". Nucleonica. Institute for Transuranium Elements. 2007–2012. Retrieved 2011-04-17.
- 1 2 3 Sonzogni, Alejandro. "Interactive Chart of Nuclides". National Nuclear Data Center: Brookhaven National Laboratory. Retrieved 4 October 2012.
- ↑ Patton, I. Jocelyn; Waldbauer, L. J. (1926). "The Radioactivity of the Alkali Metals". Chemical Reviews. 3: 81–93. doi:10.1021/cr60009a003.
- ↑ McLennan, J. C.; Kennedy, W. T. (1908). "On the radioactivity of potassium and other alkali metals". Philosophical Magazine. 6. 16 (93): 377–395. doi:10.1080/14786440908636519.
- ↑ "Potassium-40" (PDF). Human Health Fact Sheet. Argonne National Laboratory, Environmental Science Division. August 2005. Retrieved 7 February 2012.
- 1 2 Fontani, Marco (10 September 2005). "The Twilight of the Naturally-Occurring Elements: Moldavium (Ml), Sequanium (Sq) and Dor (Do)". International Conference on the History of Chemistry. Lisbon. pp. 1–8. Archived from the original on 24 February 2006. Retrieved 8 April 2007.
- 1 2 Van der Krogt, Peter (10 January 2006). "Francium". Elementymology & Elements Multidict. Retrieved 8 April 2007.
- ↑ National Institute of Standards and Technology. "Radionuclide Half-Life Measurements". Retrieved 2011-11-07.
- ↑ Radioisotope Brief: Cesium-137 (Cs-137). U.S. National Center for Environmental Health
- 1 2 The Radiological Accident in Goiânia. IAEA. 1988.
- ↑ Delacroix, D.; Guerre, J. P.; Leblanc, P.; Hickman, C. (2002). Radionuclide and Radiation Protection Data Handbook 2002 (2nd ed.). Nuclear Technology Publishing. ISBN 1-870965-87-6.
- ↑ L. Brown, Theodore; LeMay, Jr., H. Eugene; Bursten, Bruce E.; Burdge, Julia R. (2003). Chemistry: The Central Science (8th ed.). US: Pearson Education. ISBN 0-13-061142-5.
- 1 2 Clark, Jim (2005). "Reaction of the Group 1 Elements with Water". chemguide. Retrieved 18 June 2012.
- ↑ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "Electronegativity".
- ↑ Goldberg, David E. (1988). 3,000 Solved Problems in Chemistry (1st ed.). McGraw-Hill. ISBN 0-07-023684-4. Section 17.43, page 321
- ↑ Theodore, Louis; Dupont, R. Ryan; Ganesan, Kumar, eds. (1999). Pollution Prevention: The Waste Management Approach to the 21st Century. CRC Press. p. 15 Section 27. ISBN 1-56670-495-2.
- 1 2 Clark, Jim (2000). "Metallic Bonding". chemguide. Retrieved 23 March 2012.
- 1 2 "Coulomb explosion during the early stages of the reaction of alkali metals with water". Nature Chemistry. 7: 250–254. 26 Jan 2015. doi:10.1038/nchem.2161.
- ↑ Buszek, Keith R. (2001) "Sodium Amalgam" in Encyclopedia of Reagents for Organic Synthesis, Wiley. doi:10.1002/047084289X.rs040
- ↑ "Sodium-Potassium Alloy (NaK)" (PDF). BASF.
- ↑ Kaner, Richard (2003). "Cesium". Chemical and Engineering News.
- ↑ Sevov, S.C. "Zintl Phases", pp. 113–132 in Intermetallic Compounds, Principles and Practice: Progress, Vol. 3. Westbrook, J.H.; Freisher, R.L.: Eds.; John Wiley & Sons. Ltd., Chichester, England doi:10.1002/0470845856 ISBN 978-0-470-84585-1
- 1 2 S.M. Kauzlarich, Encyclopedia of Inorganic chemistry, 1994, John Wiley & Sons, ISBN 0-471-93620-0
- ↑ Hagen, A. P. (17 September 2009). Inorganic Reactions and Methods, The Formation of Bonds to Group-I, -II, and -IIIB Elements. John Wiley & Sons. pp. 204–5. ISBN 978-0-470-14549-4.
- ↑ Matkovich, V. I. (6 December 2012). Boron and Refractory Borides. Springer. pp. 262–92. ISBN 978-3-642-66620-9.
- ↑ Hermann, Andreas; McSorley, Alexandra; N. W., Ashcroft; Hoffmann, Roald (2012). "From Wade–Mingos to Zintl–Klemm at 100 GPa: Binary Compounds of Boron and Lithium" (PDF). Journal of the American Chemical Society. 2012 (134): 18606–18. doi:10.1021/ja308492g. Retrieved 21 August 2016.
- ↑ Housecroft, Catherine E.; Sharpe, Alan G. (2008). "Chapter 14: The group 14 elements". Inorganic Chemistry, 3rd Edition. Pearson. p. 386. ISBN 978-0-13-175553-6.
- ↑ NIST Ionizing Radiation Division 2001 – Technical Highlights. physics.nist.gov
- ↑ Emery, N.; et al. (2008). "Review: Synthesis and superconducting properties of CaC6". Sci. Technol. Adv. Mater. 9 (4): 044102. Bibcode:2008STAdM...9d4102E. PMC 5099629
 . PMID 27878015. doi:10.1088/1468-6996/9/4/044102.
. PMID 27878015. doi:10.1088/1468-6996/9/4/044102. - ↑ Hoch, Constantin; Wendorff, Marco; Röhr, Caroline (2002). "Tetrapotassium nonastannide, K4Sn9". Acta Crystallographica Section C. 58 (4): i45. doi:10.1107/S0108270102002032.
- ↑ Gregory, Duncan H.; O'Meara, Paul M.; Gordon, Alexandra G.; Hodges, Jason P.; Short, Simine; Jorgensen, James D. (2002). "Structure of Lithium Nitride and Transition-Metal-Doped Derivatives, Li3−x−yMxN (M= Ni, Cu): A Powder Neutron Diffraction Study". Chem. Mater. 14 (5): 2063–2070. doi:10.1021/cm010718t.
- ↑ Fischer, D.; Jansen, M. (2002). "Synthesis and structure of Na3N". Angew Chem. 41 (10): 1755–1756. doi:10.1002/1521-3773(20020517)41:10<1755::AID-ANIE1755>3.0.CO;2-C.
- ↑ Fischer, D.; Cancarevic, Z.; Schön, J. C.; Jansen, M. Z. (2004). "Synthesis and structure of K3N". Z. Anorg. Allg. Chem. 630 (1): 156–160. doi:10.1002/zaac.200300280.. 'Elusive Binary Compound Prepared' Chemical & Engineering News 80 No. 20 (20 May 2002)
- ↑ H.G. Von Schnering, W. Hönle Phosphides – Solid-state Chemistry Encyclopedia of Inorganic Chemistry Ed. R. Bruce King (1994) John Wiley & Sons ISBN 0-471-93620-0
- ↑ Kahlenberg, Louis (2008). Outlines of Chemistry – A Textbook for College Students. READ BOOKS. pp. 324–325. ISBN 1-4097-6995-X.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 King, R. Bruce (1995). Inorganic Chemistry of Main Group Elements. Wiley-VCH. ISBN 0-471-18602-3.
- ↑ "Welcome to Arthur Mar's Research Group". University of Alberta. University of Alberta. 1999–2013. Retrieved 24 June 2013.
- ↑ Lindsay, D. M.; Garland, D. A. (1987). "ESR spectra of matrix-isolated lithium superoxide". The Journal of Physical Chemistry. 91 (24): 6158–61. doi:10.1021/j100308a020.
- ↑ Vol'nov, I. I.; Matveev, V. V. (1963). "Synthesis of cesium ozonide through cesium superoxide". Bulletin of the Academy of Sciences, USSR Division of Chemical Science. 12 (6): 1040–1043. doi:10.1007/BF00845494.
- ↑ Tokareva, S. A. (1971). "Alkali and Alkaline Earth Metal Ozonides". Russian Chemical Reviews. 40 (2): 165–174. Bibcode:1971RuCRv..40..165T. doi:10.1070/RC1971v040n02ABEH001903.
- ↑ Simon, A. (1997). "Group 1 and 2 Suboxides and Subnitrides — Metals with Atomic Size Holes and Tunnels". Coordination Chemistry Reviews. 163: 253–270. doi:10.1016/S0010-8545(97)00013-1.
- ↑ Tsai, Khi-Ruey; Harris, P. M.; Lassettre, E. N. (1956). "The Crystal Structure of Tricesium Monoxide". Journal of Physical Chemistry. 60 (3): 345–347. doi:10.1021/j150537a023.
- ↑ Okamoto, H. (2009). "Cs-O (Cesium-Oxygen)". Journal of Phase Equilibria and Diffusion. 31: 86–87. doi:10.1007/s11669-009-9636-5.
- ↑ Band, A.; Albu-Yaron, A.; Livneh, T.; Cohen, H.; Feldman, Y.; Shimon, L.; Popovitz-Biro, R.; Lyahovitskaya, V.; Tenne, R. (2004). "Characterization of Oxides of Cesium". The Journal of Physical Chemistry B. 108 (33): 12360–12367. doi:10.1021/jp036432o.
- ↑ Brauer, G. (1947). "Untersuchungen ber das System Cäsium-Sauerstoff". Zeitschrift für anorganische Chemie. 255: 101–124. doi:10.1002/zaac.19472550110.
- ↑ Butterman, William C.; Brooks, William E.; Reese, Jr., Robert G. (2004). "Mineral Commodity Profile: Cesium" (PDF). United States Geological Survey. Archived from the original (PDF) on 7 February 2007. Retrieved 27 December 2009.
- ↑ House, James E. (2008). Inorganic chemistry. Academic Press. p. 524. ISBN 0-12-356786-6.
- ↑ Moyer, Harvey V. (1956). "Chemical Properties of Polonium". In Moyer, Harvey V. Polonium. Oak Ridge, Tenn.: United States Atomic Energy Commission. pp. 33–96. doi:10.2172/4367751. TID-5221.
- ↑ Bagnall, K. W. (1962). "The Chemistry of Polonium". Adv. Inorg. Chem. Radiochem. Advances in Inorganic Chemistry and Radiochemistry. 4: 197–229. ISBN 978-0-12-023604-6. doi:10.1016/S0065-2792(08)60268-X.
- ↑ Alberto, R.; Ortner, K.; Wheatley, N.; Schibli, R.; Schubiger, A. P. (2001). "Synthesis and properties of boranocarbonate: a convenient in situ CO source for the aqueous preparation of [99mTc(OH2)3(CO)3]+". J. Am. Chem. Soc. 121 (13): 3135–3136. doi:10.1021/ja003932b.
- ↑ Cotton, F. A.; Wilkinson, G. (1972). Advanced Inorganic Chemistry. John Wiley and Sons Inc. ISBN 0-471-17560-9.
- ↑ T. Kottke, D. Stalke (September 1993). "Structures of Classical Reagents in Chemical Synthesis: (nBuLi)6, (tBuLi)4, and the Metastable (tBuLi · Et2O)2". Angew. Chem., Int. Ed. Engl. 32 (4): 580–582. doi:10.1002/anie.199305801.
- ↑ Elschenbroich, C. "Organometallics" (2006) Wiley-VCH: Weinheim. ISBN 3-527-29390-6.
- ↑ Dinnebier, R. E.; Behrens, U.; Olbrich, F. (1998). "Lewis Base-Free Phenyllithium: Determination of the Solid-State Structure by Synchrotron Powder Diffraction". Journal of the American Chemical Society. 120 (7): 1430–1433. doi:10.1021/ja972816e.
- ↑ Brown, T. L.; Rogers, M. T. (1957). "The Preparation and Properties of Crystalline Lithium Alkyls". Journal of the American Chemical Society. 79 (8): 1859–1861. doi:10.1021/ja01565a024.
- ↑ Schlosser, Manfred (1988). "Superbases for organic synthesis". Pure and Appl. Chem. 60 (11): 1627–1634. doi:10.1351/pac198860111627.
- ↑ Clegg, William; Conway, Ben; Kennedy, Alan R.; Klett, Jan; Mulvey, Robert E.; Russo, Luca (2011). "Synthesis and Structures of \(Trimethylsilyl)methyl]sodium and -potassium with Bi- and Tridentate N-Donor Ligands". European Journal of Inorganic Chemistry. 2011 (5): 721–726. doi:10.1002/ejic.201000983.
- 1 2 3 4 5 Pyykkö, Pekka (2011). "A suggested periodic table up to Z ≤ 172, based on Dirac–Fock calculations on atoms and ions". Physical Chemistry Chemical Physics. 13 (1): 161–8. Bibcode:2011PCCP...13..161P. PMID 20967377. doi:10.1039/c0cp01575j.
- 1 2 3 Seaborg, G. T. (c. 2006). "transuranium element (chemical element)". Encyclopædia Britannica. Retrieved 16 March 2010.
- ↑ Gäggeler, Heinz W. (5–7 November 2007). "Gas Phase Chemistry of Superheavy Elements" (PDF). Lecture Course Texas A&M. Archived from the original (PDF) on 20 February 2012. Retrieved 26 February 2012.
- ↑ Fricke, Burkhard (1975). "Superheavy elements: a prediction of their chemical and physical properties". Recent Impact of Physics on Inorganic Chemistry. 21: 89–144. doi:10.1007/BFb0116498. Retrieved 4 October 2013.
- ↑ Kratz, J. V. (5 September 2011). The Impact of Superheavy Elements on the Chemical and Physical Sciences (PDF). 4th International Conference on the Chemistry and Physics of the Transactinide Elements. Retrieved 27 August 2013.
- ↑ Nuclear scientists eye future landfall on a second 'island of stability'. EurekAlert! (2008-04-06). Retrieved on 2016-11-25.
- ↑ Grumann, Jens; Mosel, Ulrich; Fink, Bernd; Greiner, Walter (1969). "Investigation of the stability of superheavy nuclei around Z=114 and Z=164". Zeitschrift für Physik. 228 (5): 371–386. doi:10.1007/BF01406719.
- 1 2 3 Dietzel, P. D.; Kremer, R. K.; Jansen, M. (8 January 2007). "Superoxide compounds of the large pseudo-alkali-metal ions tetramethylammonium, -phosphonium, and -arsonium.". Chem Asian J. 2 (1): 66–75. PMID 17441140. doi:10.1002/asia.200600306.
- 1 2 "International Union of Pure and Applied Chemistry > Periodic Table of the Elements". IUPAC. Retrieved 1 May 2011.
- ↑ Folden, Cody (31 January 2009). "The Heaviest Elements in the Universe" (PDF). Saturday Morning Physics at Texas A&M. Archived from the original (PDF) on 10 August 2014. Retrieved 9 March 2012.
- ↑ Emsley, J. (1989). The Elements. Oxford: Clarendon Press. pp. 22–23.
- ↑ Winter, Mark J. (1994) Chemical Bonding, Oxford University Press, ISBN 0-19-855694-2
- 1 2 3 Vinson, Greg (2008). "Hydrogen is a Halogen". HydrogenTwo.com. Archived from the original on 10 January 2012. Retrieved 14 January 2012.
- 1 2 3 4 5 6 Cronyn, Marshall W. (August 2003). "The Proper Place for Hydrogen in the Periodic Table" (PDF). Journal of Chemical Education. 80 (8): 947–951. Bibcode:2003JChEd..80..947C. doi:10.1021/ed080p947.
- ↑ Huheey, J.E.; Keiter, E.A. and Keiter, R.L. (1993) Inorganic Chemistry: Principles of Structure and Reactivity, 4th edition, HarperCollins, New York, USA.
- ↑ James, A.M. and Lord, M.P. (1992) Macmillan's Chemical and Physical Data, Macmillan, London, UK.
- ↑ Wigner, E.; Huntington, H. B. (1935). "On the possibility of a metallic modification of hydrogen". Journal of Chemical Physics. 3 (12): 764. Bibcode:1935JChPh...3..764W. doi:10.1063/1.1749590.
- ↑ Nellis, W. J.; Weir, S. T.; Mitchell, A. C. (1999). "Metallization of fluid hydrogen at 140 GPa (1.4 Mbar) by shock compression". Shock Waves. 9: 301–305. Bibcode:1999ShWav...9..301N. doi:10.1007/s001930050189.
- ↑ Cousins, David M.; Davidson, Matthew G.; García-Vivó, Daniel (2013). "Unprecedented participation of a four-coordinate hydrogen atom in the cubane core of lithium and sodium phenolates". Chem. Commun. 49: 11809–11811. doi:10.1039/c3cc47393g. Retrieved 7 August 2014.
- ↑ Leach, Mark R. "2002 Inorganic Chemist's Periodic Table". Retrieved 16 October 2012.
- ↑ Holleman, A. F.; Wiberg, E. (2001), Inorganic Chemistry, San Diego: Academic Press, ISBN 0-12-352651-5
- 1 2 3 Stevenson, D. J. (20 November 1975). "Does metallic ammonium exist?". Nature. Nature Publishing Group. 258 (5532): 222–223. Bibcode:1975Natur.258..222S. doi:10.1038/258222a0.
- 1 2 Bernal, M. J. M.; Massey, H. S. W. (3 February 1954). "Metallic Ammonium". Monthly Notices of the Royal Astronomical Society. Wiley-Blackwell for the Royal Astronomical Society. 114: 172–179. Bibcode:1954MNRAS.114..172B. doi:10.1093/mnras/114.2.172.
- ↑ "Solubility Rules!". chem.sc.edu.
- ↑ Reedy, J. H. (October 1, 1929). "Lecture demonstration of ammonium amalgam". Journal of Chemical Education. 6 (10): 1767. doi:10.1021/ed006p1767.
- ↑ Connelly, Neil G.; Geiger, William E. (1996). "Chemical Redox Agents for Organometallic Chemistry". Chemical Reviews. 96 (2): 877–910. PMID 11848774. doi:10.1021/cr940053x.
- ↑ El Murr, N.; Sheats, J. E.; Geiger, W. E.; Holloway, J. D. L. (1979). "Electrochemical Reduction Pathways of the Rhodocenium Ion. Dimerization and Reduction of Rhodocene". Inorg. Chem. 18 (6): 1443–1446. doi:10.1021/ic50196a007.
- ↑ Keller, H. J.; Wawersik, H. (1967). "Spektroskopische Untersuchungen an Komplexverbindungen. VI. EPR-spektren von (C5H5)2Rh und (C5H5)2Ir". J. Organomet. Chem. (in German). 8 (1): 185–188. doi:10.1016/S0022-328X(00)84718-X.
- ↑ Shannon, R. D. (1976). "Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides". Acta Crystallogr A. 32 (5): 751–767. Bibcode:1976AcCrA..32..751S. doi:10.1107/S0567739476001551.
- 1 2 Crookes, William (1864). "On Thallium". The Journal of the Chemical Society, London. Harrison & Sons. 17: 112–152. doi:10.1039/js8641700112. Retrieved 13 January 2012.
- 1 2 3 4 Leach, Mark R. (1999–2012). "The Internet Database of Periodic Tables". meta-synthesis.com. Retrieved 6 April 2012.
- ↑ International Union of Pure and Applied Chemistry (2005). Nomenclature of Inorganic Chemistry (IUPAC Recommendations 2005). Cambridge (UK): RSC–IUPAC. ISBN 0-85404-438-8. pp. 51. Electronic version..
- ↑ Mudring, Anja-Verena (2007). "Thallium Halides – New Aspects of the Stereochemical Activity of Electron Lone Pairs of Heavier Main-Group Elements". European Journal of Inorganic Chemistry. 2007 (6): 882–890. doi:10.1002/ejic.200600975.
- ↑ Russell AM & Lee KL (2005) Structure-property relations in nonferrous metals. Wiley-Interscience, New York. p. 302. ISBN 0-471-64952-X
- ↑ Deming HG (1940) Fundamental Chemistry, John Wiley & Sons, New York, pp. 705–7
- 1 2 3 4 5 Jensen, William B. (2003). "The Place of Zinc, Cadmium, and Mercury in the Periodic Table" (PDF). Journal of Chemical Education. American Chemical Society. 80 (8): 952–961. Bibcode:2003JChEd..80..952J. doi:10.1021/ed080p952. Archived from the original (PDF) on 11 June 2010. Retrieved 2012-05-06.
- ↑ Bailar, J. C. (1973) Comprehensive inorganic chemistry, vol. 3, p. 16. ISBN 1-57215-291-5
- ↑ "Salary". Retrieved 20 July 2015.
- ↑ Marggraf, Andreas Siegmund (1761). Chymische Schriften. p. 167.
- ↑ du Monceau, H. L. D. "Sur la Base de Sel Marine". Memoires de l'Academie royale des Sciences (in French): 65–68.
- 1 2 Weeks, Mary Elvira (1932). "The discovery of the elements. IX. Three alkali metals: Potassium, sodium, and lithium". Journal of Chemical Education. 9 (6): 1035. Bibcode:1932JChEd...9.1035W. doi:10.1021/ed009p1035.
- 1 2 Siegfried, R. (1963). "The Discovery of Potassium and Sodium, and the Problem of the Chemical Elements". Isis. 54 (2): 247–258. JSTOR 228541. doi:10.1086/349704.
- ↑ Enghag, P. (2004). "11. Sodium and Potassium". Encyclopedia of the elements. Wiley-VCH Weinheim. ISBN 3-527-30666-8.
- 1 2 Davy, Humphry (1808). "On some new phenomena of chemical changes produced by electricity, in particular the decomposition of the fixed alkalies, and the exhibition of the new substances that constitute their bases; and on the general nature of alkaline bodies". Philosophical Transactions of the Royal Society of London. 98: 1–44. doi:10.1098/rstl.1808.0001 .
- ↑ Shaposhnik, V. A. (2007). "History of the discovery of potassium and sodium (on the 200th anniversary of the discovery of potassium and sodium)". Journal of Analytical Chemistry. 62 (11): 1100–1102. doi:10.1134/S1061934807110160.
- ↑ Ralph, Jolyon; Chau, Ida (24 August 2011). "Petalite: Petalite mineral information and data". Retrieved 27 November 2011.
- 1 2 Winter, Mark. "WebElements Periodic Table of the Elements | Lithium | historical information". Retrieved 27 November 2011.
- ↑ Weeks, Mary (2003). Discovery of the Elements. Whitefish, Montana, United States: Kessinger Publishing. p. 124. ISBN 0-7661-3872-0. Retrieved 10 August 2009.
- ↑ "Johan Arfwedson". Archived from the original on 5 June 2008. Retrieved 10 August 2009.
- 1 2 van der Krogt, Peter. "Lithium". Elementymology & Elements Multidict. Retrieved 5 October 2010.
- ↑ Clark, Jim (2005). "Compounds of the Group 1 Elements". chemguide. Retrieved 10 August 2009.
- ↑ Kaner, Richard (2003). "C&EN: It's Elemental: The Periodic Table – Cesium". American Chemical Society. Retrieved 25 February 2010.
- ↑ Kirchhoff, G.; Bunsen, R. (1861). "Chemische Analyse durch Spectralbeobachtungen". Annalen der Physik und Chemie. 189 (7): 337–381. Bibcode:1861AnP...189..337K. doi:10.1002/andp.18611890702.
- ↑ Weeks, Mary Elvira (1932). "The discovery of the elements. XIII. Some spectroscopic discoveries". Journal of Chemical Education. 9 (8): 1413–1434. Bibcode:1932JChEd...9.1413W. doi:10.1021/ed009p1413.
- ↑ Oxford English Dictionary, 2nd Edition
- ↑ Newlands, John A. R. (20 August 1864). "On Relations Among the Equivalents". Chemical News. 10: 94–95. Archived from the original on 1 January 2011. Retrieved November 25, 2013.
- ↑ Newlands, John A. R. (18 August 1865). "On the Law of Octaves". Chemical News. 12: 83. Archived from the original on 1 January 2011. Retrieved November 25, 2013.
- ↑ Mendelejew, Dimitri (1869). "Über die Beziehungen der Eigenschaften zu den Atomgewichten der Elemente". Zeitschrift für Chemie (in German): 405–406.
- ↑ Fluck, E. (1988). "New Notations in the Periodic Table" (PDF). Pure Appl. Chem. IUPAC. 60 (3): 431–436. doi:10.1351/pac198860030431. Retrieved November 25, 2013.
- ↑ "Alabamine & Virginium". TIME. 15 February 1932. Retrieved 1 April 2007.
- ↑ MacPherson, H. G. (1934). "An Investigation of the Magneto-Optic Method of Chemical Analysis". Physical Review. American Physical Society. 47 (4): 310–315. Bibcode:1935PhRv...47..310M. doi:10.1103/PhysRev.47.310.
- ↑ Adloff, Jean-Pierre; Kaufman, George B. (2005-09-25). Francium (Atomic Number 87), the Last Discovered Natural Element Archived 4 June 2013 at the Wayback Machine.. The Chemical Educator 10 (5). Retrieved on 26 March 2007.
- ↑ "Francium". McGraw-Hill Encyclopedia of Science & Technology. 7. McGraw-Hill Professional. 2002. pp. 493–494. ISBN 0-07-913665-6.
- 1 2 Lougheed, R. W.; Landrum, J. H.; Hulet, E. K.; Wild, J. F.; Dougan, R. J.; Dougan, A. D.; Gäggeler, H.; Schädel, M.; Moody, K. J.; Gregorich, K. E.; Seaborg, G. (1985). "Search for superheavy elements using 48Ca + 254Esg reaction". Physical Review C. 32 (5): 1760–1763. Bibcode:1985PhRvC..32.1760L. doi:10.1103/PhysRevC.32.1760.
- ↑ van der Krogt, Peter. "Ununennium". Elementymology & Elements Multidict. Retrieved 14 February 2011.
- ↑ Schadel, M.; Brüchle, W.; Brügger, M.; Gäggeler, H.; Moody, K.; Schardt, D.; Sümmerer, K.; Hulet, E.; Dougan, A.; et al. (1986). "Heavy isotope production by multinucleon transfer reactions with 254Es". Journal of the Less Common Metals. 122: 411–417. doi:10.1016/0022-5088(86)90435-2.
- ↑ "Modern alchemy: Turning a line". The Economist. 12 May 2012. Retrieved 5 October 2012.
- ↑ Emsley, John (2011). Nature's Building Blocks: An A-Z Guide to the Elements (New ed.). New York, NY: Oxford University Press. p. 593. ISBN 978-0-19-960563-7.
- 1 2 Lodders, Katharina (2003). "Solar System Abundances and Condensation Temperatures of the Elements". The Astrophysical Journal. 591 (2): 1220–1247. Bibcode:2003ApJ...591.1220L. doi:10.1086/375492 .
- ↑ Oddo, Giuseppe (1914). "Die Molekularstruktur der radioaktiven Atome". Zeitschrift für anorganische Chemie. 87: 253–268. doi:10.1002/zaac.19140870118.
- ↑ Harkins, William D. (1917). "The Evolution of the Elements and the Stability of Complex Atoms. I. A New Periodic System Which Shows a Relation Between the Abundance of the Elements and the Structure of the Nuclei of Atoms". Journal of the American Chemical Society. 39 (5): 856–879. doi:10.1021/ja02250a002.
- ↑ North, John (2008). Cosmos an illustrated history of astronomy and cosmology (Rev. and updated ed.). Univ. of Chicago Press. p. 602. ISBN 978-0-226-59441-5.
- ↑ Morgan, J. W.; Anders, E. (1980). "Chemical composition of Earth, Venus, and Mercury". Proceedings of the National Academy of Sciences. 77 (12): 6973–6977. Bibcode:1980PNAS...77.6973M. PMC 350422 . PMID 16592930. doi:10.1073/pnas.77.12.6973.
- ↑ Albarède, Francis (2003). Geochemistry: an introduction. Cambridge University Press. ISBN 978-0-521-89148-6.
- ↑ "Abundance in Earth's Crust". WebElements.com. Retrieved 14 April 2007.
- ↑ "List of Periodic Table Elements Sorted by Abundance in Earth's crust". Israel Science and Technology Homepage. Retrieved 15 April 2007.
- 1 2 3 Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ↑ "Lithium Occurrence". Institute of Ocean Energy, Saga University, Japan. Archived from the original on 2 May 2009. Retrieved 13 March 2009.
- ↑ "Some Facts about Lithium". ENC Labs. Retrieved 15 October 2010.
- ↑ Schwochau, Klaus (1984). "Extraction of metals from sea water". Topics in Current Chemistry. Topics in Current Chemistry. 124/1984: 91–133. ISBN 978-3-540-13534-0. doi:10.1007/3-540-13534-0_3.
- ↑ Wise, M. A. (1995). "Trace element chemistry of lithium-rich micas from rare-element granitic pegmatites". Mineralogy and Petrology. 55 (13): 203–215. Bibcode:1995MinPe..55..203W. doi:10.1007/BF01162588.
- ↑ CRC Handbook of Chemistry and Physics. 4. CRC. 2006. p. 12. ISBN 0-8493-0474-1.
- 1 2 3 Emsley, John (2001). Nature's Building Blocks. Oxford: Oxford University Press. pp. 151–153. ISBN 0-19-850341-5.
- 1 2 Gagnon, Steve. "Francium". Jefferson Science Associates, LLC. Archived from the original on 31 March 2007. Retrieved 1 April 2007.
- 1 2 Winter, Mark. "Geological information". Francium. The University of Sheffield. Retrieved 26 March 2007.
- ↑ "It's Elemental — The Periodic Table of Elements". Jefferson Lab. Archived from the original on 29 April 2007. Retrieved 14 April 2007.
- ↑ Ober, Joyce A. "Lithium" (PDF). United States Geological Survey. pp. 77–78. Archived (PDF) from the original on 11 July 2007. Retrieved 19 August 2007.
- ↑ Pauling, Linus. General Chemistry (1970 ed.). Dover Publications.
- ↑ "Los Alamos National Laboratory – Sodium". Retrieved 8 June 2007.
- ↑ Merck Index, 9th ed., monograph 8325
- ↑ Winter, Mark. "WebElements Periodic Table of the Elements | Potassium | Essential information". Webelements. Retrieved 27 November 2011.
- ↑ Lemke, Charles H.; Markant, Vernon H. (2001). "Sodium and Sodium Alloys". Kirk-Othmer Encyclopedia of Chemical Technology. ISBN 0-471-23896-1. doi:10.1002/0471238961.1915040912051311.a01.pub2.
- ↑ "Cesium and Rubidium Hit Market". Chemical & Engineering News. 37 (22): 50–56. 1959. doi:10.1021/cen-v037n022.p050.
- 1 2 3 4 Butterman, William C.; Brooks, William E.; Reese, Jr., Robert G. (2003). "Mineral Commodity Profile: Rubidium" (PDF). United States Geological Survey. Retrieved 4 December 2010.
- ↑ bulletin 585. United States. Bureau of Mines. 1995.
- ↑ Burt, R. O. (1993). "Caesium and cesium compounds". Kirk-Othmer encyclopedia of chemical technology. 5 (4th ed.). New York: John Wiley & Sons, Inc. pp. 749–764. ISBN 978-0-471-48494-3.
- ↑ Stancari, G.; Veronesi, S.; Corradi, L.; Atutov, S. N.; Calabrese, R.; Dainelli, A.; Mariotti, E.; Moi, L.; Sanguinetti, S.; Tomassetti, L. (2006). "Production of Radioactive Beams of Francium". Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 557 (2): 390–396. Bibcode:2006NIMPA.557..390S. doi:10.1016/j.nima.2005.11.193.
- 1 2 Orozco, Luis A. (2003). "Francium". Chemical and Engineering News.
- 1 2 Price, Andy (December 20, 2004). "Francium". Retrieved February 19, 2012.
- ↑ USGS (2011). "Lithium" (PDF). Retrieved 4 December 2011.
- ↑ "Soaps & Detergents: Chemistry". Retrieved 20 July 2015.
- ↑ Lindsey, Jack L (1997). Applied illumination engineering. p. 112. ISBN 978-0-88173-212-2.
- ↑ Kane, Raymond; Sell, Heinz (2001). Revolution in lamps: A chronicle of 50 years of progress. p. 241. ISBN 978-0-88173-351-8.
- ↑ Stampers, National Association of Drop Forgers and (1957). Metal treatment and drop forging.
- ↑ Harris, Jay C (1949). Metal cleaning bibliographical abstracts. p. 76.
- ↑ Cordel, Oskar (1868). Die Stassfurter Kalisalze in der Landwirthschalt: Eine Besprechung ... (in German). L. Schnock. Retrieved 29 May 2011.
- ↑ Toedt, John; Koza, Darrell; Cleef-Toedt, Kathleen Van (2005). "Personal Cleansing Products: Bar Soap". Chemical composition of everyday products. Greenwood Publishing Group. ISBN 978-0-313-32579-3.
- ↑ Schultz, H.; et al. (2006). "Potassium compounds". Ullmann's Encyclopedia of Industrial Chemistry. A22. p. 95. ISBN 3-527-30673-0. doi:10.1002/14356007.a22_031.pub2.
- ↑ "Cesium Atoms at Work". Time Service Department—U.S. Naval Observatory—Department of the Navy. Archived from the original on 23 February 2015. Retrieved 20 December 2009.
- ↑ "The NIST reference on Constants, Units, and Uncertainty". National Institute of Standards and Technology.
- ↑ Koch, E.-C. (2002). "Special Materials in Pyrotechnics, Part II: Application of Caesium and Rubidium Compounds in Pyrotechnics". Journal Pyrotechnics. 15: 9–24.
- ↑ Heiserman, David L. (1992). Exploring Chemical Elements and their Compounds. McGraw-Hill. pp. 201–203. ISBN 0-8306-3015-5.
- ↑ Winter, Mark. "Uses". Francium. The University of Sheffield. Archived from the original on 31 March 2007. Retrieved 25 March 2007.
- ↑ Gomez, E.; Orozco, L. A.; Sprouse, G. D. (7 November 2005). "Spectroscopy with trapped francium: advances and perspectives for weak interaction studies". Rep. Prog. Phys. 69 (1): 79–118. Bibcode:2006RPPh...69...79G. doi:10.1088/0034-4885/69/1/R02.
- ↑ Peterson, I. (11 May 1996). "Creating, cooling, trapping francium atoms" (PDF). Science News. 149 (19): 294. doi:10.2307/3979560. Retrieved 11 September 2009.
- 1 2 3 Lerner, Michael M. (2013). "Standard Operating Procedure: Storage and Handling of Alkali Metals". Oregon State University. Retrieved 26 August 2016.
- ↑ Solomon, Robert E. (2002). Fire and Life Safety Inspection Manual. Jones & Bartlett Learning. p. 459. ISBN 978-0-87765-472-8.
- ↑ Angelici, R. J. (1999). Synthesis and Technique in Inorganic Chemistry. Mill Valley, CA: University Science Books. ISBN 0-935702-48-2.
- ↑ Martel, Bernard; Cassidy, Keith (2004-07-01). "Rubidium". Chemical risk analysis: a practical handbook. p. 215. ISBN 978-1-903996-65-2.
- ↑ Wray, Thomas K. "Danger: peroxidazable chemicals" (PDF). Environmental Health & Public Safety (North Carolina State University).
- ↑ Astrid, Sigel; Helmut, Sigel; Roland K.O., Sigel, eds. (2016). The Alkali Metal Ions: Their Role in Life. Metal Ions in Life Sciences. 16. Springer. doi:10.1007/978-3-319-21756-7.
- ↑ Katsuyuki, Aoki; Kazutaka, Murayama; Hu, Ning-Hai (2016). "Chapter 3. Solid State Structuresof Alkali Metal Ion Complexes Formed by Low-Molecular-Weight Ligands of Biological Relevance". In Astrid, Sigel; Helmut, Sigel; Roland K.O., Sigel. The Alkali Metal Ions: Their Role in Life. Metal Ions in Life Sciences. 16. Springer. pp. 27–101. doi:10.1007/978-3-319-21756-7_3.
- 1 2 3 4 5 Winter, Mark. "WebElements Periodic Table of the Elements | Lithium | biological information". Webelements. Retrieved 15 February 2011.
- ↑ Gray, Theodore. "Facts, pictures, stories about the element Lithium in the Periodic Table". theodoregray.com. Retrieved 9 January 2012.
- ↑ Howland, Robert H. (September 2007). "Lithium: Underappreciated and Underused?". Psychiatric Annals. 37 (9). Retrieved 6 November 2012.
- ↑ Zarse, Kim; Terao, Takeshi; Tian, Jing; Iwata, Noboru; Ishii, Nobuyoshi; Ristow, Michael (August 2011). "Low-dose lithium uptake promotes longevity in humans and metazoans". European Journal of Nutrition. 50 (5): 387–9. PMC 3151375 . PMID 21301855. doi:10.1007/s00394-011-0171-x.
- 1 2 3 Winter, Mark. "WebElements Periodic Table of the Elements | Potassium | biological information". WebElements. Retrieved 13 January 2012.
- 1 2 Winter, Mark. "WebElements Periodic Table of the Elements | Sodium | biological information". WebElements. Retrieved 13 January 2012.
- ↑ "Sodium" (PDF). Northewestern University. Archived from the original (PDF) on 23 August 2011. Retrieved 21 November 2011.
- ↑ "Sodium and Potassium Quick Health Facts". Retrieved 7 November 2011.
- ↑ "Dietary Reference Intakes: Water, Potassium, Sodium, Chloride, and Sulfate". Food and Nutrition Board, Institute of Medicine, United States National Academies. 11 February 2004. Retrieved 23 November 2011.
- ↑ U.S. Department of Agriculture; U.S. Department of Health and Human Services (December 2010). Dietary Guidelines for Americans, 2010 (PDF) (7th ed.). p. 22. ISBN 978-0-16-087941-8. OCLC 738512922. Archived from the original (PDF) on 27 October 2011. Retrieved 23 November 2011.
- ↑ Geleijnse, J. M.; Kok, F. J.; Grobbee, D. E. (2004). "Impact of dietary and lifestyle factors on the prevalence of hypertension in Western populations". European Journal of Public Health. 14 (3): 235–239. PMID 15369026. doi:10.1093/eurpub/14.3.235 .
- ↑ Lawes, C. M.; Vander Hoorn, S.; Rodgers, A.; International Society of Hypertension (2008). "Global burden of blood-pressure-related disease, 2001" (PDF). Lancet. 371 (9623): 1513–1518. PMID 18456100. doi:10.1016/S0140-6736(08)60655-8. Archived from the original (PDF) on 28 January 2012.
- 1 2 Hellgren, Mikko; Sandberg, Lars; Edholm, Olle (2006). "A comparison between two prokaryotic potassium channels (KirBac1.1 and KcsA) in a molecular dynamics (MD) simulation study". Biophys. Chem. 120 (1): 1–9. PMID 16253415. doi:10.1016/j.bpc.2005.10.002.
- 1 2 Schonwald, Seth (2004). "Potassium Chloride and Potassium Permanganate". Medical toxicology. Lippincott Williams & Wilkins. pp. 903–5. ISBN 978-0-7817-2845-4.
- ↑ Markovchick, Vincent J. & Pons, Peter T. (2003). Emergency medicine secrets. Elsevier Health Sciences. p. 223. ISBN 978-1-56053-503-4.
- 1 2 3 Winter, Mark. "WebElements Periodic Table of the Elements | Rubidium | biological information". Webelements. Retrieved 15 February 2011.
- 1 2 Relman, A. S. (1956). "The Physiological Behavior of Rubidium and Cesium in Relation to That of Potassium". The Yale Journal of Biology and Medicine. 29 (3): 248–62. PMC 2603856 . PMID 13409924.
- 1 2 Meltzer, H. L. (1991). "A pharmacokinetic analysis of long-term administration of rubidium chloride". Journal of clinical pharmacology. 31 (2): 179–84. PMID 2010564. doi:10.1002/j.1552-4604.1991.tb03704.x.
- 1 2 Winter, Mark. "WebElements Periodic Table of the Elements | Caesium | biological information". WebElements. Retrieved 13 January 2012.
- ↑ Fieve, Ronald R.; Meltzer, Herbert L.; Taylor, Reginald M. (1971). "Rubidium chloride ingestion by volunteer subjects: Initial experience". Psychopharmacologia. 20 (4): 307–14. PMID 5561654. doi:10.1007/BF00403562.
- ↑ Meltzer, H. L. (1991). "A pharmacokinetic analysis of long-term administration of rubidium chloride". Journal of clinical pharmacology. 31 (2): 179–84. PMID 2010564. doi:10.1002/j.1552-4604.1991.tb03704.x.
- ↑ Follis, Richard H., Jr. (1943). "Histological Effects in rats resulting from adding Rubidium or Cesium to a diet deficient in potassium". AJP: Legacy. 138 (2): 246.
- ↑ Gottschlich, Michele M. (2001). The Science and Practice of Nutrition Support: A Case-based Core Curriculum. Kendall Hunt. p. 98. ISBN 978-0-7872-7680-5. Retrieved 9 July 2016.
- ↑ Insel, Paul M.; Turner, R. Elaine; Ross, Don (2004). Nutrition. Jones & Bartlett Learning. p. 499. ISBN 978-0-7637-0765-1. Retrieved 10 July 2016.
- ↑ Melnikov, P.; Zanoni, L. Z. (June 2010). "Clinical effects of cesium intake". Biological trace element research. 135 (1–3): 1–9. PMID 19655100. doi:10.1007/s12011-009-8486-7.
- ↑ Pinsky, Carl; Bose, Ranjan; Taylor, J. R.; McKee, Jasper; Lapointe, Claude; Birchall, James (1981). "Cesium in mammals: Acute toxicity, organ changes and tissue accumulation". Journal of Environmental Science and Health, Part A. 16 (5): 549– 567. doi:10.1080/10934528109375003.
- ↑ Pinsky, Carl; Bose, Ranjan; Taylor, J. R.; McKee, Jasper; Lapointe, Claude; Birchall, James (1981). "Cesium in mammals: Acute toxicity, organ changes and tissue accumulation". Journal of Environmental Science and Health, Part A. 16 (5): 549– 567. doi:10.1080/10934528109375003.
- ↑ Johnson, Garland T.; Lewis, Trent R.; Wagner, D. Wagner (1975). "Acute toxicity of cesium and rubidium compounds". Toxicology and Applied Pharmacology. 32 (2): 239–245. PMID 1154391. doi:10.1016/0041-008X(75)90216-1.
- ↑ Sartori, H. E. (1984). "Cesium therapy in cancer patients". Pharmacol Biochem Behav. 21 (Suppl 1): 11–13. PMID 6522427. doi:10.1016/0091-3057(84)90154-0.
- ↑ Wood, Leonie. "'Cured' cancer patients died, court told". The Sydney Morning Herald. 20 November 2010.
| Periodic table forms |
| ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sets of elements |
| ||||||||||||||||||
| Elements | |||||||||||||||||||
| History | |||||||||||||||||||
| See also | |||||||||||||||||||
| |||||||||||||||||||