XXXY syndrome
| XXXY syndrome | |
|---|---|
| Classification and external resources | |
| ICD-10 | Q98.1 |
| Orphanet | 96263 |
XXXY syndrome is a sex chromosome aneuploidy in which males have two extra X chromosomes.[1] Human cells usually inherit two sex chromosomes, one from the mother and one from the father. Usually, females have two X chromosomes (XX) and males have one X and one Y chromosome (XY). The appearance of at least one Y chromosome with a properly functioning SRY gene makes a male. As a result, XXXY only affects males. Males affected with XXXY syndrome have 48 chromosomes instead of the typical 46. This is why XXXY syndrome is sometimes referred to as 48, XXXY syndrome or 48, XXXY. The symptoms of this condition is very similar to Klinefelter syndrome, including delayed or reduced male puberty and infertility. It is estimated that XXXY affects one in every 50,000 male births..
Non-Invasive Prenatal Testing (NIPT) can give a positive result for XXXY syndrome in the fetus. This test assumes the mother has two X chromosomes (XX). Since the occurrence of XXXY syndrome is very rare, it is more likely that the mother has Triple X syndrome (or XXX). In this case, doctors or genetic counselors should recommend amniocentensis or CVS to determine the sex of the fetus.
Signs and symptoms
The symptoms of 48, XXXY syndrome are similar to those of Klinefelter syndrome, though usually the symptoms are more severe in 48, XXXY syndrome. Like Klinefelter syndrome, the presence of additional X chromosomes affects the male reproductive system, can cause physical abnormalities, and can affect cognitive development. There is a greater risk for congenetial malformations and more medical problems.
Reproductive
Males with XXXY syndrome can have testicular dysgenesis and hypergonadotrophic hypogonadism.[2] Testicular dygenesis is a condition in which a male with 48, XXXY is born with that results in infertility as a result of incomplete or complete loss of spermatogenesis, therefore the male is unable to produce sperm or produces low levels of sperm. Hypergonadotrophic hypogonadism is a condition in which the function of the testes in males is reduced and can result in low levels of sex steroids produced like testosterone.
Physical
Males with 48, XXXY have a tall stature, which becomes more prominent in adulthood. Facial dysmorphism is common in males with 48, XXXY and can include increased distance between the eyes (hypertelorism), skin folds of the upper eyelid (epicanthal folds), up-slanting opening between the eyelids (palpebral fissures) and hooded eyelids. Other physical features include the fifth finger or "pinky" to be bent inwards towards the fourth finger (clinodactyly), short nail beds, flat feet, double jointedness (hyperextensibility) and prominent elbows with cubitus varus where the arm rests closer to the body. Musculoskeletal features may include congentical elbow dislocation and the limited ability of the feet to roll inwards while walking and upon landing.[2]
Cognitive and developmental
Neurological effects are believed to be more severe as the number of extra X chromosomes increases; a male with 48, XXXY is likely to have more severe symptoms than a male with Klinefelter syndrome. Developmental delays are common in infancy and childhood. Expected symptoms include speech delays, motor delays, and hypotonia (lack of muscle tone), also known as floppy baby syndrome. Cognitive abilities tend to decrease 10-15 IQ points with each additional X chromosome and the IQ score of a male with 48, XXXY can range from 20 to 78. Other neurodevelopmental and psychological disorders are also symptoms of 48, XXXY syndrome and the severity is often more severe than in a male with Klinefelter syndrome, 47, XXY. Social development and behavior may also be affected in an individual with 48, XXXY.[3]
Cause
The cause of 48, XXXY can be from non-disjunction in the paternal sperm or non-disjunction in the maternal oocyte.[2] In the case where the sperm is the genetic cause of 48, XXXY syndrome, the sperm would have to contain two X chromosomes and one Y chromosome. This would be caused by two non-disjunction events in spermatogenesis, both meosis I and meosis II. The duplicated X chromosome in the sperm would have to fail to separate in both meosis I and meosis II for a sperm as well as the X and Y chromosomes would have to be in the same sperm. Then the XXY sperm would fertilize a normal oocyte to make a XXXY zygote. In the case where the oocyte is the genetic cause of 48, XXXY syndrome the oocyte would contain three X chromosome. This would be caused by two non-disjunction events during oogenesis. In meosis I both sets of duplicated X chromosomes would have to be not separated. Then in meosis II one set of X chromosomes would have to not separate and the other set would separate resulting in one oocyte with three X chromosomes. A normal sperm containing a Y chromosome would have to fertilize the XXX oocyte to make a XXXY zygote.[4]
Diagnosis
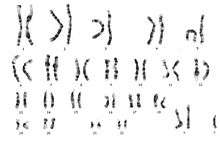
Diagnosis of 48, XXXY is usually done by a standard karyotype.[2] A karyotype is a chromosomal analysis in which a full set of chromosomes can be seen for an individual. In a male with 48, XXXY the diagnosis would be confirmed by finding one Y chromosome and three X chromosomes therefore two extra X chromosomes.
Another way to diagnosis 48, XXXY is by chromosomal microarray showing the presence of extra X chromosomes.[2] Chromosomal microarray (CMA) is used to detect extra or missing chromosomal segments or whole chromosomes. CMA uses microchip-based testing to analyze many pieces of DNA. The CMA chips use labels or probes to bind to specific chromosome regions. An individual with 48, XXXY would have extra X chromosome regions detected by CMA, indicating multiple X chromosomes.Males with 48, XXXY are diagnosed anywhere from before birth to adulthood as a result of the range in the severity of symptoms.[2]
See also
References
- ↑ "48,XXXY syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 2017-03-20.
- 1 2 3 4 5 6 Tartaglia, Nicole; Ayari, Natalie; Howell, Susan; D’Epagnier, Cheryl; Zeitler, Philip (2011-06-01). "48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome". Acta Paediatrica. 100 (6): 851–860. ISSN 1651-2227. PMC 3314712
 . PMID 21342258. doi:10.1111/j.1651-2227.2011.02235.x.
. PMID 21342258. doi:10.1111/j.1651-2227.2011.02235.x. - ↑ Gropman A, Samango-Sprouse CA. 2013. Neurocognitive variance and neurological underpinnings of the X and Y chromosomal variations. Am J Med Genet Part C Semin Med Genet 163C: 35–43.
- ↑ Greenstein, Robert M.; Harris, David J.; Luzzatti, Luigi; Cann, Howard M. (1970-04-01). "Cytogenetic Analysis of a Boy with the Xxxy Syndrome: Origin of the X-Chromosomes". Pediatrics. 45 (4): 677–686. ISSN 0031-4005. PMID 5438170.
Further reading
- Ferrier, Pierre E. (1974-01-01). "The XXXY Klinefelter Syndrome in Childhood". Archives of Pediatrics & Adolescent Medicine. 127 (1). ISSN 0002-922X. doi:10.1001/archpedi.1974.02110200106016.