Zero differential overlap
Zero differential overlap is an approximation in computational molecular orbital theory that is the central technique of semi-empirical methods in quantum chemistry. When computers were first used to calculate bonding in molecules, it was possible to only calculate diatomic molecules. As computers advanced, it became possible to study larger molecules, but the use of this approximation has always allowed the study of even larger molecules. Currently semi-empirical methods can be applied to molecules as large as whole proteins. The approximation involves ignoring certain integrals, usually two-electron repulsion integrals. If the number of orbitals used in the calculation is N, the number of two-electron repulsion integrals scales as N4. After the approximation is applied the number of such integrals scales as N2, a much smaller number, simplifying the calculation.
Details of approximation
If the molecular orbitals 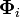 are expanded in terms of N basis functions,
are expanded in terms of N basis functions, 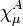 as:-
as:-
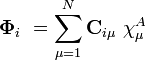
where A is the atom the basis function is centred on, and 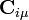 are coefficients, the two-electron repulsion integrals are then defined as:-
are coefficients, the two-electron repulsion integrals are then defined as:-
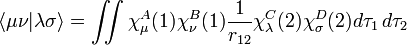
The zero differential overlap approximation ignores integrals that contain the product 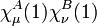 where μ is not equal to ν. This leads to:-
where μ is not equal to ν. This leads to:-
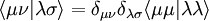
where 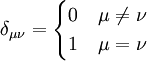
The total number of such integrals is reduced to N(N + 1) / 2 (approximately N2 / 2) from [N(N + 1) / 2][N(N + 1) / 2 + 1] / 2 (approximately N4 / 8), all of which are included in ab initio Hartree–Fock and post-Hartree–Fock calculations.
Scope of approximation in semi-empirical methods
Methods such as the Pariser–Parr–Pople method (PPP) and CNDO/2 use the zero differential overlap approximation completely. Methods based on the intermediate neglect of differential overlap, such as INDO, MINDO, ZINDO and SINDO do not apply it when A = B = C = D, i.e. when all four basis functions are on the same atom. Methods that use the neglect of diatomic differential overlap, such as MNDO, PM3 and AM1, also do not apply it when A = B and C = D, i.e. when the basis functions for the first electron are on the same atom and the basis functions for the second electron are the same atom.
It is possible to partly justify this approximation, but generally it is used because it works reasonably well when the integrals that remain – 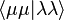 – are parameterised.
– are parameterised.
References
- Jensen, Frank (1999). Introduction to Computational Chemistry. Chichester: John Wiley and Sons. pp. 81–82. ISBN 978-0-471-98085-8. OCLC 466189317.