Woodward–Hoffmann rules
The Woodward–Hoffmann rules, devised by Robert Burns Woodward and Roald Hoffmann, are a set of rules in organic chemistry predicting the barrier heights of pericyclic reactions based upon conservation of orbital symmetry. The Woodward–Hoffmann rules can be applied to understand electrocyclic reactions, cycloadditions (including cheletropic reactions), sigmatropic reactions, and group transfer reactions. Reactions are classified as allowed if the electronic barrier is low, and forbidden if the barrier is high. Forbidden reactions can still take place but require significantly more energy.
The Woodward–Hoffmann rules were first formulated to explain the striking stereospecificity of electrocyclic reactions under thermal and photochemical control. Thermolysis of the substituted cyclobutene trans-1,2,3,4-tetramethylcyclobutene (1) gave only one geometric isomer, the (E,E)-3,4-dimethyl-2,4-hexadiene (2) as shown below; the (Z,Z) and the (E,Z) geometric isomers were not detected in the reaction. Similarly, thermolysis of cis-1,2,3,4-tetramethylcyclobutene (3) gave only the (E,Z) geometric isomer (4).[1]

Due to their elegance and simplicity, the Woodward–Hoffmann rules are credited with first exemplifying the power of molecular orbital theory to experimental chemists.[2] Hoffmann was awarded the 1981 Nobel Prize in Chemistry for this work, shared with Kenichi Fukui who developed a similar model using frontier molecular orbital (FMO) theory; because Woodward had died two years before, he was not eligible to win what would have been his second Nobel Prize for Chemistry.[3]
Original formulation
The Woodward–Hoffmann rules were first invoked to explain the observed stereospecificity of electrocyclic ring-opening and ring-closing reactions at the termini of open chain conjugated polyenes either by application of heat (thermal reactions) or application of light (photochemical reactions).
In the original publication in 1965,[4] the three rules distilled from experimental evidence and molecular orbital analysis appeared as follows:
- In an open-chain system containing 4n-electrons, the orbital symmetry of the highest occupied molecule orbital is such that a bonding interaction between the termini must involve overlap between orbital envelopes on opposite faces of the system and this can only be achieved in a conrotatory process.
- In open systems containing 4n + 2 electrons, terminal bonding interaction within ground-state molecules requires overlap of orbital envelopes on the same face of the system, attainable only by disrotatory displacements.
- In a photochemical reaction an electron in the HOMO of the reactant is promoted to an excited state leading to a reversal of terminal symmetry relationships and reversal of stereospecificity.
Using this formulation it is possible to understand the stereospecifity of the electrocyclic ring-closure of the substituted 1,3-butadiene pictured below. 1,3-butadiene has 4  -electrons in the ground state and thus proceeds through a conrotatory ring-closing mechanism.
-electrons in the ground state and thus proceeds through a conrotatory ring-closing mechanism.

Conversely in the electrocyclic ring-closure of the substituted 1,3,5-hexatriene pictured below, the reaction proceeds through a disrotatory mechanism.

In the case of a photochemically driven electrocyclic ring-closure of 1,3-butadiene, electronic promotion causes  becomes the HOMO and the reaction mechanism must be disrotatory.
becomes the HOMO and the reaction mechanism must be disrotatory.
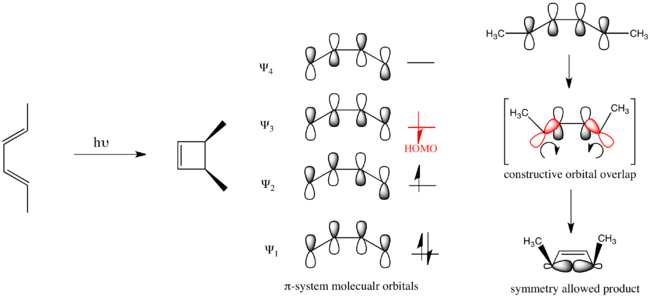
Organic reactions that obey these rules are said to be symmetry allowed. Reactions that take the opposite course are symmetry forbidden and require substantially more energy to take place if they take place at all.
Correlation diagrams
The Woodward–Hoffmann rules can also be derived by examining the correlation diagram of a given reaction.[5][6] A symmetry element is a point of reference (usually a plane or a line) about which an object is symmetric with respect to a symmetry operation. If a symmetry element is present throughout the reaction mechanism (reactant, transition state, and product), it is called a conserved symmetry element. Then, throughout the reaction, the symmetry of molecular orbitals with respect to this element must be conserved. That is, molecular orbitals that are symmetric with respect to the symmetry element in the starting material must be correlated to (transform into) orbitals symmetric with respect to that element in the product. Conversely, the same statement holds for antisymmetry with respect to a conserved symmetry element. A molecular orbital correlation diagram correlates molecular orbitals of the starting materials and the product based upon conservation of symmetry. From a molecular orbital correlation diagram one can construct an electronic state correlation diagram that correlates electronic states (i.e. ground state, and excited states) of the reactants with electronic states of the products. Correlation diagrams can then be used to predict the height of transition state barriers.[7]
Electrocyclic reactions

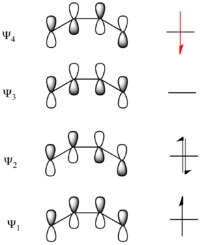
Considering the electrocyclic ring closure of the substituted 1,3-butadiene, the reaction can proceed through either a conrotatory or a disrotatory reaction mechanism. As shown to the left, in the conrotatory transition state there is a C2 axis of symmetry and in the disrotatory transition state there is a σ mirror plane of symmetry. In order to correlate orbitals of the starting material and product, one must determine whether the molecular orbitals are symmetric or antisymmetric with respect to these symmetry elements. The π-system molecular orbitals of butadiene are shown to the right along with the symmetry element with which they are symmetric. They are antisymmetric with respect to the other. For example, Ψ2 of 1,3-butadiene is symmetric with respect to 180o rotation about the C2 axis, and antisymmetric with respect to reflection in the mirror plane.

Ψ1 and Ψ3 are symmetric with respect to the mirror plane as the sign of the p-orbital lobes is preserved under the symmetry transformation. Similarly, Ψ1 and Ψ3 are antisymmetric with respect to the C2 axis as the rotation inverts the sign of the p-orbital lobes uniformly. Conversely Ψ2 and Ψ4 are symmetric with respect to the C2 axis and antisymmetric with respect to the σ mirror plane.

The same analysis can be carried out for the molecular orbitals of cyclobutene. The result of both symmetry operations on each of the MOs is shown to the left. Note that here as the σ and σ* orbitals lie entirely in the plane containing C2 perpendicular to σ, they are uniformly symmetric and antisymmetric (respectively) to both symmetry elements. On the other hand, π is symmetric with respect to reflection and antisymmetric with respect to rotation, while π* is antisymmetric with respect to reflection and symmetric with respect to rotation.
Correlation lines are drawn to connect molecular orbitals in the starting material and the product that have the same symmetry with respect to the conserved symmetry element. In the case of the conrotatory 4 electron electrocyclic ring closure of 1,3-butadiene, the lowest molecular orbital Ψ1 is asymmetric (A) with respect to the C2 axis. So this molecular orbital is correlated with the π orbital of cyclobutene, the lowest energy orbital that is also (A) with respect to the C2 axis. Similarly, Ψ2, which is symmetric (S) with respect to the C2 axis, is correlated with σ of cyclobutene. The final two correlations are between the antisymmetric (A) molecular orbitals Ψ3 and σ*, and the symmetric (S) molecular orbitals Ψ4 and π*.[6]

Similarly, there exists a correlation diagram for a disrotatory mechanism. In this mechanism, the symmetry element that persists throughout the entire mechanism is the σ mirror plane of reflection. Here the lowest energy MO Ψ1 of 1,3-butadiene is symmetric with respect to the reflection plane, and as such correlates with the symmetric σ MO of cyclobutene. Similarly the higher energy pair of symmetric molecular orbitals Ψ3 and π correlate. As for the asymmetric molecular orbitals, the lower energy pair Ψ2 and π* form a correlation pair, as do Ψ4 and σ*.[6]

Evaluating the two mechanisms, the conrotatory mechanism is predicted to have a lower barrier because it transforms the electrons from ground-state orbitals of the reactants (Ψ1 and Ψ2) into ground-state orbitals of the product (σ and π). Conversely, the disrotatory mechanism forces the conversion of the Ψ1 orbital into the σ orbital, and the Ψ2 orbital into the π* orbital. Thus the two electrons in the ground-state Ψ2 orbital are transferred to an excited antibonding orbital, creating a doubly excited electronic state of the cyclobutene. This would lead to a significantly higher transition state barrier to reaction.[6]

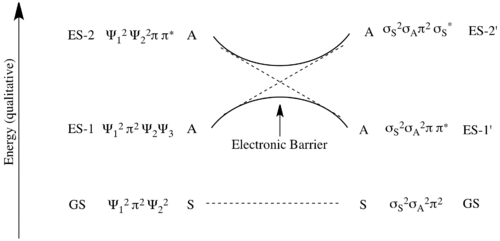
However, as reactions do not take place between disjointed molecular orbitals, but electronic states, the final analysis involves state correlation diagrams. A state correlation diagram correlates the overall symmetry of electronic states in the starting material and product. The ground state of 1,3-butadiene, as shown above, has 2 electrons in Ψ1 and 2 electrons in Ψ2, so it is represented as Ψ12Ψ22. The overall symmetry of the state is the product of the symmetries of each filled orbital with multiplicity for doubly populated orbitals. Thus, as Ψ1 is asymmetric with respect to the C2 axis, and Ψ2 is symmetric, the total state is represented by A2S2. To see why this particular product is mathematically overall S, note that S can be represented as (+1) and A as (−1). This derives from the fact that signs of the lobes of the p-orbitals are multiplied by (+1) if they are symmetric with respect to a symmetry transformation (i.e. unaltered) and multiplied by (−1) if they are antisymmetric with respect to a symmetry transformation (i.e. inverted). Thus A2S2=(−1)2(+1)2=+1=S. The first excited state (ES-1) is formed from promoting an electron from the HOMO to the LUMO, and thus is represented as Ψ12Ψ2Ψ3. As Ψ1is A, Ψ2 is S, and Ψ3 is A, the symmetry of this state is given by A2SA=A.
Now considering the electronic states of the product, cyclobutene, the ground-state is given by σ2π2, which has symmetry S2A2=S. The first excited state (ES-1') is again formed from a promotion of an electron from the HOMO to the LUMO, so in this case it is represented as σ2ππ*. The symmetry of this state is S2AS=A.
The ground state Ψ12Ψ22 of 1,3-butadiene correlates with the ground state σ2π2 of cyclobutene as demonstrated in the MO correlation diagram above. Ψ1 correlates with π and Ψ2 correlates with σ. Thus the orbitals making up Ψ12Ψ22 must transform into the orbitals making up σ2π2 under a conrotatory mechanism. However, the state ES-1 does not correlate with the state ES-1' as the molecular orbitals do not transform into each other under the symmetry-requirement seen in the molecular orbital correlation diagram. Instead as Ψ1 correlates with π, Ψ2 correlates with σ, and Ψ3 correlates with σ*, the state Ψ12Ψ2Ψ3 attempts to transform into π2σσ*, which is a different excited state. So ES-1 attempts to correlate with ES-2'=σπ2σ*, which is higher in energy than Es-1'. Similarly ES-1'=σ2ππ* attempts to correlate with ES-2=Ψ1Ψ22Ψ4. These correlations can not actually take place due to the quantum-mechanical rule known as the avoided crossing rule. This says that energetic configurations of the same symmetry can not cross on an energy level correlation diagram. In short, this is caused by mixing of states of the same symmetry when brought close enough in energy. So instead a high energetic barrier is formed between a forced transformation of ES-1 into ES-1'. In the diagram below the symmetry-preferred correlations are shown in dashed lines and the bold curved lines indicate the actual correlation with the high energetic barrier.[6][7]

The same analysis can be applied to the disrotatory mechanism to create the following state correlation diagram.[6][7]

Thus if the molecule is in the ground state it will proceed through the conrotatory mechanism (i.e. under thermal control) to avoid an electronic barrier. However, if the molecule is in the first excited state (i.e. under photochemical control), the electronic barrier is present in the conrotatory mechanism and the reaction wil proceed through the disrotatory mechanism. It is important to note that these are not completely distinct as both the conrotatory and disrotatory mechanisms lie on the same potential surface. Thus a more correct statement is that as a ground state molecule explores the potential energy surface, it is more likely to achieve the activation barrier to undergo a conrotatory mechanism.[7]
Cycloaddition reactions
The Woodward–Hoffmann rules can also explain bimolecular cycloaddition reactions through correlation diagram analysis.[8] A [πp+πq] cycloaddition brings together two components, one with p π-electrons, and the other with q π-electrons. Cycloaddition reactions are further characterized as suprafacial (s) or antarafacial (a) with respect to each of the π components.
[2+2] cycloadditions
There are 4 possible mechanisms for the [2+2] cycloadditon: [π2s+π2s], [π2a+π2s], [π2s+π2a], [π2a+π2a]. However, in reality, the only reaction that, in general, can proceed due to geometric constraints is suprafacial with respect to both components.[6]
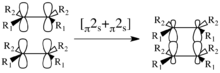
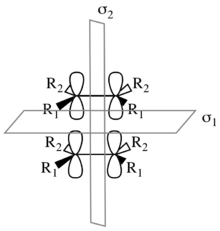
Considering the [π2s+π2s] cycloaddition. This mechanism leads to a retention of stereochemistry in the product, as illustrated to the right. There are two symmetry elements present in the starting materials, transition state, and product: σ1 and σ2. σ1 is the mirror plane between the components perpendicular to the p-orbitals; σ2 splits the molecules in half perpendicular to the σ-bonds.[8] Note that these are both local-symmetry elements in the case that the components are not identical.

To determine symmetry and asymmetry with respect to σ1 and σ2, the starting material molecular orbitals must be considered in tandem. The figure to the right shows the molecular orbital correlation diagram for the [π2s+π2s] cycloaddition. The two π and π* molecular orbitals of the starting materials are characterized by their symmetry with respect to first σ1 and then σ2. Similarly, the σ and σ* molecular orbitals of the product are characterized by their symmetry. In the correlation diagram, molecular orbitals transformations over the course of the reaction must conserve the symmetry of the molecular orbitals. Thus πSS correlates with σSS, πAS correlates with σ*AS, π*SA correlates with σSA, and finally π*AA correlates with σ*AA. Due to conservation of orbital symmetry, the bonding orbital πAS is forced to correlate with the antibonding orbital σ*AS. Thus a high barrier is predicted.[6][7][8]
This is made precise in the state correlation diagram below.[6][7] The ground state in the starting materials is the electronic state where πSS and πAS are both doubly populated – i.e. the state (SS)2(AS)2. As such, this state attempts to correlate with the electronic state in the product where both σSS and σ*AS are doubly populated – i.e. the state (SS)2(AS)2. However, this state is neither the ground state (SS)2(SA)2 of cyclobutane, nor the first excited state ES-1'=(SS)2(SA)(AS), where an electron is promoted from the HOMO to the LUMO. So the ground state of the reactants attempts to correlate with a second excited state ES-2'=(SS)2(AS)2.
Similarly, the ground state of the product cyclobutane, as can be seen in the molecular orbital diagram above, is the electronic state where both σSS and σSA are doubly populated – i.e. the state (SS)2(SA)2. This attempts to correlate with the state where πSS and π*SA are both doubly populated – i.e. a second excited state ES-2=(SS)2(SA)2.
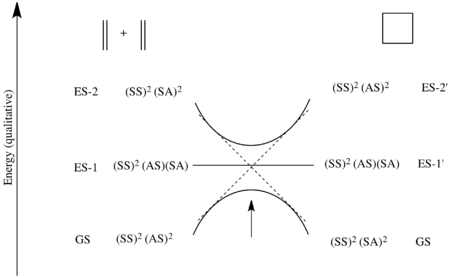
Finally, the first excited state of the starting materials is the electronic configuration where πSS is doubly populated, and πAS and π*SA are both singly occupied – i.e. the state (SS)2(AS)(SA). The first excited state of the product is also the state (SS)2(SA)(AS) as σSS is doubly populated, and σSA and σ*AS are both singly occupied. Thus these two excited states correlate.
The ground state of the starting materials only attempts to correlate with the second excited state as there is an avoided crossing in the middle due to the states possessing the overall same symmetry. Thus in actuality, the ground state of the reactants is transformed into the ground state of the products only after achieving a high energetic barrier. However, there is no large activation barrier if the reactants are in the first excited state. Thus this reaction proceeds easily under photo-control, but has a very high barrier to reaction under thermal control.
[4+2] cycloadditions
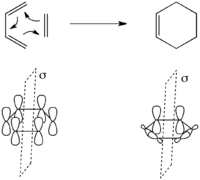
A [π4+π2] cycloaddition is a concerted 2-component pericyclic reaction exemplified by the Diels-Alder reaction. The simplest case is the reaction of 1,3-butadiene with ethylene to form cyclohexene shown to the left.
There is only one conserved symmetry element in this transformation – the mirror plane through the center of the reactants as shown to the left. From this we can assign the symmetry of the molecular orbitals of the reactants very simply. The molecular orbitals of the reactants are merely the set {Ψ1, Ψ2, Ψ3, Ψ4} of molecular orbitals of 1,3-butadiene shown above, along with π and π* of ethylene. Ψ1 is symmetric, Ψ2 is antisymmetric, Ψ3 is symmetric, and Ψ4 is symmetric with respect to the mirror plane. Similarly π is symmetric and π* is antisymmetric with respect to the mirror plane.

The molecular orbitals of the product are the symmetric and antisymmetric combinations of the two newly formed σ and σ* bonds and the π and π* bonds as shown below.
Correlating the pairs of orbitals in the starting materials and product of the same symmetry and increasing energy gives the correlation diagram to the right. As this transforms the ground state bonding molecular orbitals of the starting materials into the ground state bonding orbitals of the product in a symmetry conservative manner this is predicted to not have the great energetic barrier present in the ground state [2+2] reaction above.
To make the analysis precise, one can construct the state correlation diagram for the general [4+2]-cycloaddition.[7] As before, the ground state is the electronic state depicted in the molecular orbital correlation diagram to the right. This can be described as Ψ12π2Ψ22, of total symmetry S2S2 A2=S. This correlates with the ground state of the cyclohexene σSσAπ2 which is also S2S2A2=S. As such this ground state reaction is not predicted to have a high symmetry-imposed barrier.
One can also construct the excited-state correlations as is done above. Here, there is a high energetic barrier to a photo-induced Diels-Alder reaction under a suprafacial-suprafacial bond topology due to the avoided crossing shown below.
Group transfer reactions
The symmetry-imposed barrier heights of group transfer reactions can also be analyzed using correlation diagrams. A model reaction is the transfer of a pair of hydrogen atoms from ethane to perdeuterioethane shown to the right.
The only conserved symmetry element in this reaction is the mirror plane through the center of the molecules as shown to the left.
The molecular orbitals of the system are constructed as symmetric and antisymmetic combinations of σ and σ* C–H bonds in ethane, and π and π* bonds in the deutero-substituted ethylene. Thus the lowest energy MO is the symmetric sum of the two C–H σ-bond (σS), followed by the antisymmetric sum (σA). The two highest energy MOs are formed from linear combinations of the σCH antibonds – highest is the antisymmetric σ*A, preceded by the symmetric σ*A at a slightly lower energy. In the middle of the energetic scale are the two remaining MOs that are the πCC and π*CC of ethylene.
The full molecular orbital correlation diagram is constructed in by matching pairs of symmetric and asymmetric MOs of increasing total energy, as explained above. As can be seen in the diagram to the right, as the bonding orbitals of the reactants exactly correlate with the bonding orbitals of the products this reaction is not predicted to have a high electronic symmetry-imposed barrier.[6][7]
Selection rules
Using correlation diagrams one can derive selection rules for the following generalized classes of pericyclic reactions. Each of these particular classes is further generalized in the generalized Woodward–Hoffmann rules. The more inclusive bond topology descriptors antarafacial and suprafacial subsume the terms conrotatory and disrotatory, respectively. Antarafacial refers to bond making or breaking through the opposite face of a π system, p orbital, or σ bond, while suprafacial refers to the process occurring through the same face. A suprafacial transformation at a chiral center preserves stereochemistry, whereas an antarafacial transformation reverses stereochemistry.
Electrocyclic reactions
The selection rule of electrocyclization reactions is given in the original statement of the Woodward–Hoffmann rules. If a generalized electrocyclic ring closure occurs in a polyene of 4n π-electrons, then it is conrotatory under thermal conditions and disrotatory under photochemical conditions. Conversely in a polyene of 4n+2 π-electrons, an electrocyclic ring closure is disrotatory under thermal conditions and conrotatory under photochemical conditions.
This result can either be derived via an FMO analysis based upon the sign of p-orbital lobes of the HOMO of the polyene or with correlation diagrams. Taking first the first possibility, in the ground state, if a polyene has 4n electrons, the outer p-orbitals of the HOMO that form the σ bond in the electrocyclized product are of opposite signs. Thus a constructive overlap is only produced under a conrotatory/antarafacial process. Conversely for a polyene with 4n+2 electrons, the outer p-orbitals of the ground state HOMO are of the same sign. Thus constructive orbital overlap occurs with a disrotatory/suprafacical process.[4]

Additionally, the correlation diagram for any 4n electrocyclic reaction will resemble the diagram for the 4 electron cyclization of 1,3-butadiene, while the correlation diagram any 4n+2 electron electrocyclic reaction will resemble the correlation diagram for the 6 electron cyclization of 1,3,5-hexatriene.[6]
This is summarized in the following table:
| Thermally allowed | Photochemically allowed | |
|---|---|---|
| 4n | conrotatory | disrotatory |
| 4n+2 | disrotatory | conrotatory |
Sigmatropic rearrangement reactions
A general sigmatropic rearrangement can be classified as order [i,j], meaning that a σ bond originally between atoms denoted 1 and 1', adjacent to one or more π systems, is shifted to between atoms i and j. Thus it migrates (i−1), (j−1) atoms away from its original position.
Correlation diagrams are of no use in analyzing the conservation of orbital symmetry in sigmatropic rearrangements as there are, in general, only symmetry elements present in the transition state – i.e. no conserved symmetry elements.[6][7] Thus most useful analysis comes from FMO theory.

One of the most prevalent classes of sigmatropic shifts is classified as [1,j], where j is odd. That means a σ-bond migrates (j-1) bonds away across a π-system in a reaction involving j+1 electrons: j-1 from the π-system and 2 from σ-bond. Using FMO analysis, [1,j]-sigmatropic rearrangements are allowed if the transition state has constructive overlap between the migrating group and the accepting p-orbital of the HOMO. In [1,j]-sigmatropic rearrangements if 1+j=4n, then supra/antara is thermally allowed, and if 1+j = 4n+2, then supra/supra or antara/antara is thermally allowed.[7]
The other prevalent class of sigmatropic rearrangements are [3,3], notably the Cope and Claisen rearrangements. Here, the constructive interactions must be between the HOMOs of the two allyl radical fragments in the transition state. The ground state HOMO Ψ2 of the allyl fragment is shown below. As the terminal p-orbitals are of opposite sign, this reaction can either take place in a supra/supra topology, or an antara/antara topology.[7]

The selection rules for an [i,j]-sigmatropic rearrangement are as follows:
- For supra/supra or antara/antara [i,j]-sigmatropic shifts, if i+j = 4n + 2 they are thermally allowed and if i + j = 4n they are photochemically allowed
- For supra/antara [i,j]-sigmatropic shifts, if i + j = 4n they are thermally allowed, and if i+j = 4n + 2 they are photochemically allowed
This is summarized in the following table:
| i+j | Thermally allowed | Photochemically allowed |
|---|---|---|
| 4n | is +ja or ia +js | is +js or ia +ja |
| 4n+2 | is +js or ia +ja | is +ja or ia +js |
Cycloaddition reactions
A general [p + q]-cycloaddition is a concerted addition reaction between two components, one with p π-electrons, and one with q π-electrons. This reaction is symmetry allowed under the following conditions:[6]
- For a supra/supra or antara/antara cycloaddition, it is thermally allowed if p + q = 4n + 2 and photochemically allowed if p+q=4n
- For a supra/antara cycloaddition, it is thermally allowed if p+q=4n and photochemically allowed if p + q = 4n + 2
This is summarized in the following table:
| p+q | Thermally allowed | Photochemically allowed |
|---|---|---|
| 4n | ps +qa or pa +qs | ps +qs or pa +qa |
| 4n + 2 | ps +qs or pa +qa | ps +qa or pa +qs |
Group transfer reactions
A general double group transfer reaction which is synchronous can be represented as an interaction between a component with p π electrons and a component with q π electrons as shown.
Then the selection rules are the same as for the generalized cycloaddition reactions.[6] That is
- For supra/supra or antara/antara double group transfers, if p + q = 4n + 2 it is thermally allowed, and if p + q = 4n it is photochemically allowed
- For supra/antara double group transfers, if p + q = 4n it is thermally allowed, and if p + q = 4n + 2 it is photochemically allowed
This is summarized in the following table:
| p+q | Thermally allowed | Photochemically allowed |
|---|---|---|
| 4n | ps +qa or pa +qs | ps +qs or pa +qa |
| 4n + 2 | ps +qs or pa +qa | ps +qa or pa +qs |
General formulation
Though the Woodward–Hoffmann rules were first stated in terms of electrocyclic processes, they were soon generalized to all pericyclic reactions, as the similarity in the above selection rules should indicate.

The first thing to note is that in the Generalized Woodward–Hoffmann rules, everything is characterized in terms of antarafacial and suprafacial bond topologies. A conrotatory reaction mechanism corresponds to an antarafacial bond topology as illustrated in the diagram to the right. The p-orbital lobes that make contact in the forming σ-bond are from opposite sides of the π-system. Conversely, a disrotatory mechanism corresponds to a suprafacial bond topology as the forming σ-bond is between p-orbital lobes on the same side of the π-system.
Any pericyclic reaction can be treated of as a concerted intramolecular cycloaddition reaction. Using this generalization, Woodward and Hoffmann state in their 1969 review the general formulation for all pericyclic reactions as follows:
A ground-state pericyclic change is symmetry-allowed when the total number of (4q+2)s and (4r)a components is odd.[6]
Here, (4q + 2)s and (4r)a refer to suprafacial (4q + 2)-electron and antarafacial (4r)-electron components, respectively.
Derivation of an alternative statement
Alternatively, the general statement can be formulated in terms of the total number of electrons using simple rules of divisibility. First assume that the total number of electrons is 4n + 2. Then we can write
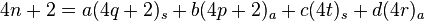
where a, b, c, and d are coefficients indicating the number of each type of component. Now, if the sum is 4n + 2 (i.e. 2 mod 4) then one of, but not both, a or b is odd. This is because if a=2k for some integer k, then
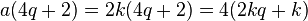
and thus 4 divides this term. However, if b=2k'+1 then
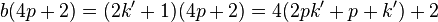
which does, in fact, leave a remainder of 2 upon division by 4. So we get the appropriate expression if one of a or b is odd and the other is even. If they were both odd then
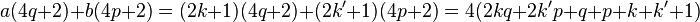
which does not leave the correct remainder upon division by 4, and thus can not be a valid expression. Thus exclusively a or b is odd.
The generalized statement of the Woodward–Hoffmann rules states that a + d is odd. Now, if a is even, then this implies that d is odd. Similarly, if a is even and the total number of electrons is 4n + 2, the derivation above shows that b must be odd. The total number of antarafacial components is b+d. If a is even, then b and d are odd so b+d is even. Now if a is odd, then d is even and b is even, so b+d is again even. Thus a 4n + 2 electron pericyclic reaction is thermally allowed if the total number of antarafacial components is even.
Considering now a 4n electron process. Now we have the conservation of electrons equation as
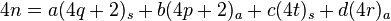
where a, b, c, and d are again the coefficients indicating the numbers of each type of component. As the entire sum is divisible by 4, a+b must be even. In other words, if a is even then b is even, and if a is odd then b is odd (i.e.  ). This is true as the above derivation for a and b of opposite parity is an if and only if statement.
). This is true as the above derivation for a and b of opposite parity is an if and only if statement.
So from the generalize Woodward–Hoffmann rules, a+d is odd. Thus if a is even, d is odd. But from the above derivation, for a 4n electron system, if a is even then b is even. Thus the total number of antarafacial components (b+d) is the sum of an even and an odd number so it is odd. Similarly if a is odd then d is even. But for a 4n electron system, if a is odd then b is also odd. Thus the total number of anatarafacial components (b+d) is again the sum of an even and an odd number and as such is odd. Thus a 4n pericyclic reaction is thermally allowed if the total number of antarafacial components is odd.
This can be summarized in the statement:
A pericyclic reaction involving 4n+2 or 4n electrons is thermally allowed if the number of antarafacial components is even or odd, respectively.
In practice, an even number of antarafacial components almost always means zero components, and an odd number almost always means one component, as transition states involving two or more antarafacial components are generally too strained to be feasible. Note that in this formulation, the electron count refers to the entire reacting system, rather than to individual components in Woodward and Hoffmann's original statement.
Examples
First, note that this formulation for a 2 component reaction is equivalent to the selection rules for a [p+q]-cycloaddition reactions shown in the following table:
| p+q | Thermally allowed | Photochemically allowed |
|---|---|---|
| 4n | ps +qa or pa +qs | ps +qs or pa +qa |
| 4n+2 | ps +qs or pa +qa | ps +qa or pa +qs |
If the total number of electrons is 4n+2, then one is in the bottom row of the table. The reaction is thermally allowed if it is suprafacial with respect to both components or antarafacial with respect to both components. That is to say the number of antarafacial components is even (it is 0 or 2). Similarly if the total number of electrons is 4n, then one is in the top row of the table. This is thermally allowed if it is suprafacial with respect to one component and antarafacial with respect to the other. Thus the total number of antarafacial components is always odd as it is always 1.
The following are some common ground state (i.e. thermal) reaction classes analyzed in light of the generalized Woodward–Hoffmann rules.
[2+2] Cycloaddition
A [2+2]-cycloaddition is a 4 electron process that brings together two components. Thus, by the above general WH rules, it is only allowed if the reaction is antarafacial with respect to exactly one component. This is the same conclusion reached with correlation diagrams in the section above.
[4+2] Cycloaddition
The synchronous 6π-electron Diels-Alder reaction is a [π4s+π2s]-cycloaddition (i.e. suprafacial with respect to both components), as exemplified by the reaction to the right.

Thus as the total number of antarafacial components is 0, which is even, the reaction is symmetry-allowed.[6] This prediction agrees with experiment as the Diels-Alder reaction is a rather facile pericyclic reaction.
4n Electrocyclic Reaction
A 4n electron electrocyclic ring opening reaction can be considered to have 2 components – the π-system and the breaking σ-bond. With respect to the π-system, the reaction is suprafacial. However, with a conrotatory mechanism, as shown in the figure above, the reaction is antarafacial with respect to the σ-bond. Conversely with a disrotatory mechanism it is suprafacial with respect to the breaking σ-bond.
By the above rules, for a 4n electron pericyclic reaction of 2 components, there must be one antarafacial component. Thus the reaction must proceed through a conrotatory mechanism.[6] This agrees with the result derived in the correlation diagrams above.
4n + 2 electrocyclic reaction
A 4n+2 electrocyclic ring opening reaction is also a 2-component pericyclic reaction which is suprafacial with respect to the π-system. Thus, in order for the reaction to be allowed, the number of antarafacial components must be 0, i.e. it must be suprafacial with respect to the breaking σ-bond as well. Thus a disrotatory mechanism is symmetry-allowed.[6]
[1,j]-sigmatropic rearrangement
A [1,j]-sigmatropic rearrangement is also a two component pericyclic reaction: one component is the π-system, the other component is the migrating group. The simplest case is a [1,j]-hydride shift across a π-system where j is odd. In this case, as the hydrogen has only a spherically symmetric s orbital, the reaction must be suprafacial with respect to the hydrogen. The total number of electrons involved is (j+1) as there are (j-1)/2 π-bond plus the σ bond involved in the reaction. If j = 4n − 1 then it must be antarafacial, and if j=4n+1, then it must be suprafacial.[6] This agrees with experiment that [1,3]-hydride shifts are generally not observed as the symmetry-allowed antarafacial process is not feasible, but [1,5]-hydride shifts are quite facile.
For a [1,j]-alkyl shift, where the reaction can be antarafacial (i.e. invert stereochemistry) with respect to the carbon center, the same rules apply. If j = 4n − 1 then the reaction is symmetry-allowed if it is either antarafacical with respect to the π-system, or inverts stereochemistry at the carbon. If j = 4n + 1 then the reaction is symmetry-allowed if it is suprafacial with respect to the π-system and retains stereochemistry at the carbon center.[6]
Equivalence of other theoretical models
Dewer–Zimmerman analysis

The generalized Woodward–Hoffmann rules stated in 1969 and 1970 are equivalent to the 1966 Möbius-Hückel concept treatment of Zimmerman.[9][10][11] In the Dewer-Zimmerman analysis, one is concerned with the topology of the transition state in the pericyclic reaction. If transition state involves 4n electrons, the Mobius topology is aromatic and allowed. If the transition state involves 4n+2 electrons, the Huckel topology is aromatic and allowed. A Mobius topology involves an odd number of phase inversions whereas a Huckel topology involves an even number of phase inversion.
Thus under any scheme of assigning relative phases to p-orbitals is acceptable as changing a phase adds an even number of inversions to the total (i.e. only the number of inversions modulo 2 is important and thus changing the phase of an orbital adds only trivial 0 modulo 2 to the total).
In the generalized Woodward-Hoffman rules, each (4r)a component has a phase inversion. With an odd number of these the transition state has a Möbius topology, and with an even number the transition state has a Hückel topology. Thus, as formulated by Woodward and Hoffmann, a 4n-electron reaction is thermally allowed if and only if it has a Mobius topology in the transition state; and a 4n + 2-electron reaction is thermally allowed if and only if it has a Huckel topology in the transition state.
Reinterpretation with conceptual density functional theory
Recently, the Woodward–Hoffmann rules have been reinterpreted using conceptual density functional theory (DFT).[2][12] The key to the analysis is the dual descriptor function[13] 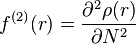 , the second derivative of the electron density
, the second derivative of the electron density  with respect to the number of electrons
with respect to the number of electrons 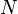 . This response function is important as the reaction of two components A and B involving a transfer of electrons will depend on the responsiveness of the electron density to electron donation or acceptance, i.e. the derivative of the Fukui function
. This response function is important as the reaction of two components A and B involving a transfer of electrons will depend on the responsiveness of the electron density to electron donation or acceptance, i.e. the derivative of the Fukui function 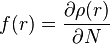 . In fact, from a simplistic viewpoint, the dual descriptor function gives a readout on the electrophilicity or nucleophilicity of the various regions of the molecule. For
. In fact, from a simplistic viewpoint, the dual descriptor function gives a readout on the electrophilicity or nucleophilicity of the various regions of the molecule. For 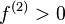 , the region is electrophilic, and for
, the region is electrophilic, and for 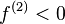 , the region is nucleophilic. Using the frontier molecular orbital assumption and a finite difference approximation of the Fukui function, one may write the dual descriptor as
, the region is nucleophilic. Using the frontier molecular orbital assumption and a finite difference approximation of the Fukui function, one may write the dual descriptor as
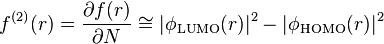
This makes intuitive sense as if a region is better at accepting electrons than donating, then the LUMO must dominate and dual descriptor function will be positive. Conversely, if a region is better at donating electrons then the HOMO term will dominate and the descriptor will be negative. Notice that although the concept of phase and orbitals are replaced simply by the notion of electron density, this function still takes both positive and negative values.

The Woodward–Hoffmann rules are reinterpreted using this formulation by matching favorable interactions between regions of electron density for which the dual descriptor has opposite signs. This is equivalent to maximizing predicted favorable interactions and minimizing repulsive interactions. For the case of a [4+2] cycloaddition, a simplified schematic of the reactants with the dual descriptor function colored (red=positive, blue=negative) is shown in the optimal supra/supra configuration to the left. This method correctly predicts the WH rules for the major classes of pericyclic reactions.
Exceptions
The Woodward–Hoffmann rules are used to predict relative barrier heights, and thus likely reaction mechanisms. It is important to remember that they only take into account barriers due to conservation of orbital symmetry. Thus it is not guaranteed that a WH symmetry-allowed reaction actually takes place in a facial manner. Conversely, it is possible, upon enough energetic input, to achieve an anti-Woodward-Hoffmann product. This is especially prevalent in sterically constrained systems, where the WH-product has an added steric barrier to overcome. For example, in the electrocyclic ring-opening of the dimethylbicyclo[0.2.3]heptene derivative (1), a conrotatory mechanism is not possible due to resulting angle strain and the reaction proceeds slowly through a disrotatory mechanism at 400o C.[4]


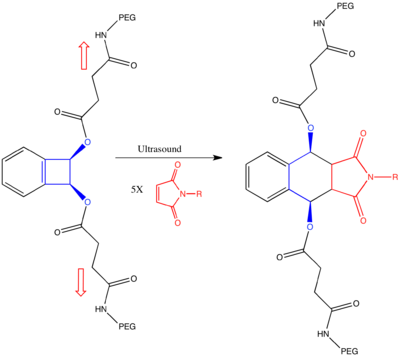
Similarly, a recent paper describes how mechanical stress can be used to reshape chemical reaction pathways to lead to products that apparently violate Woodward–Hoffman rules.[14] In this paper, they use ultrasound irradiation to induce a mechanical stress on link-functionalized polymers attached syn or anti on the cyclobutene ring. Computational studies predict that the mechanical force, resulting from friction of the polymers, induces bond lengthening along the reaction coordinate of the conrotatory mechanism in the anti-bisubstituted-cyclobutene, and along the reaction coordinate of the disrotatory mechanism in the syn-bisubstituted-cyclobutene. Thus in the syn-bisubstituted-cyclobutene, the anti-WH product is predicted to be formed.
This computational prediction was backed up by experiment on the system below. Link-functionalized polymers were conjugated to cis benzocyclobutene in both syn- and anti- conformations. As predicted, both products gave the same (Z,Z) product as determined by quenching by a stereospecific Diels-Alder reaction with the substituted maleimide. In particular, the syn-substituted product gave the anti-WH product, presumably as the mechanical stretching along the coordinate of the disrotatory pathway lowered the barrier of the reaction under the disrotatory pathway enough to bias that mechanism.
Controversy
It has been stated that Elias James Corey, also a Nobel Prize winner, feels he is responsible for the ideas that laid the foundation for this research, and that Woodward unfairly neglected to credit him in the discovery. In a 2004 memoir published in the Journal of Organic Chemistry,[15] Corey makes his claim to priority of the idea: "On May 4, 1964, I suggested to my colleague R. B. Woodward a simple explanation involving the symmetry of the perturbed (HOMO) molecular orbitals for the stereoselective cyclobutene to 1,3-butadiene and 1,3,5-hexatriene to cyclohexadiene conversions that provided the basis for the further development of these ideas into what became known as the Woodward–Hoffmann rules".
Corey, then 35, was working into the evening on Monday, May 4, as he and the other driven chemists often did. At about 8:30 p.m., he dropped by Woodward's office, and Woodward posed a question about how to predict the type of ring a chain of atoms would form. After some discussion, Corey proposed that the configuration of electrons governed the course of the reaction. Woodward insisted the solution would not work, but Corey left drawings in the office, sure that he was on to something.[16]
"I felt that this was going to be a really interesting development and was looking forward to some sort of joint undertaking," he wrote. But the next day, Woodward flew into Corey's office as he and a colleague were leaving for lunch and presented Corey's idea as his own – and then left. Corey was stunned.
In a 2004 rebuttal published in the Angewandte Chemie,[17] Roald Hoffmann denied the claim: he quotes Woodward from a lecture given in 1966 saying: "I REMEMBER very clearly—and it still surprises me somewhat—that the crucial flash of enlightenment came to me in algebraic, rather than in pictorial or geometric form. Out of the blue, it occurred to me that the coefficients of the terminal terms in the mathematical expression representing the highest occupied molecular orbital of butadiene were of opposite sign, while those of the corresponding expression for hexatriene possessed the same sign. From here it was but a short step to the geometric, and more obviously chemically relevant, view that in the internal cyclisation of a diene, the top face of one terminal atom should attack the bottom face of the other, while in the triene case, the formation of a new bond should involve the top (or pari passu, the bottom) faces of both terminal atoms."
In addition, Hoffmann points out that in two publications from 1963[18] and 1965,[19] Corey described a total synthesis of the compound dihydrocostunolide. Although they describe an electrocyclic reaction, Corey has nothing to offer with respect to explaining the stereospecificity of the synthesis.

This photochemical reaction involving 4×1+2 electrons is now recognized as conrotatory.
See also
- Woodward's rules for calculating UV absorptions
- Torquoselectivity
References
- ↑ Criegee, Rudolf; Noll, Klaus (1959). "Umsetzungen in der Reihe des 1.2.3.4-Tetramethyl-cyclobutans". Justus Liebigs Annalen der Chemie 627: 1. doi:10.1002/jlac.19596270102.
- 1 2 Geerlings, Paul; Ayers, Paul W.; Toro-Labbé, Alejandro; Chattaraj, Pratim K.; De Proft, Frank (2012). "The Woodward–Hoffmann Rules Reinterpreted by Conceptual Density Functional Theory". Accounts of Chemical Research 45 (5): 683–95. doi:10.1021/ar200192t. PMID 22283422.
- ↑ The Nobel Prize in Chemistry 1981. Nobelprize.org.
- 1 2 3 Woodward, R. B.; Hoffmann, Roald (1965). "Stereochemistry of Electrocyclic Reactions". Journal of the American Chemical Society 87 (2): 395. doi:10.1021/ja01080a054.
- ↑ Longuet-Higgins, H. C.; Abrahamson, E. W. (1965). "The Electronic Mechanism of Electrocyclic Reactions". Journal of the American Chemical Society 87 (9): 2045. doi:10.1021/ja01087a033.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 Woodward, R. B.; Hoffmann, Roald (1969). "The Conservation of Orbital Symmetry". Angew. Chem. Internat. Edit. 8 (11): 781–853. doi:10.1002/anie.196907811.
- 1 2 3 4 5 6 7 8 9 10 11 Carroll, Felix (1998) Perspectives on Structure and Mechanism in Organic Chemistry, Brooks/Cole, pp. 710–794, ISBN 0534249485.
- 1 2 3 Hoffmann, Roald; Woodward, R. B. (1965). "Selection Rules for Concerted Cycloaddition Reactions". J. Am. Chem. Soc. 87 (9): 2046. doi:10.1021/ja01087a034.
- ↑ Zimmerman, H. E. (1966). "On Molecular Orbital Correlation Diagrams, the Occurrence of Möbius Systems in Cyclization Reactions, and Factors Controlling Ground- and Excited-State Reactions. I". Journal of the American Chemical Society 88 (7): 1564. doi:10.1021/ja00959a052.
- ↑ Zimmerman, H. E. (1966). "On Molecular Orbital Correlation Diagrams, Möbius Systems, and Factors Controlling Ground and Excited State Reactions. II". J. Am. Chem. Soc. 88 (7): 1566–156. doi:10.1021/ja00959a053.
- ↑ Zimmerman, H. E. (2006). "Five decades of mechanistic and exploratory organic photochemistry". Pure Appl. Chem. 78 (12): 2193–2203. doi:10.1351/pac200678122193.
- ↑ Ayers, Paul W.; Morell, Christophe; De Proft, Frank; Geerlings, Paul (5 October 2007). "Understanding the Woodward–Hoffmann Rules by Using Changes in Electron Density". Chemistry – A European Journal 13 (29): 8240–8247. doi:10.1002/chem.200700365. Cite uses deprecated parameter
|coauthors=(help) - ↑ Morell, Christophe; Grand, André; Toro-Labbé, Alejandro (1 January 2005). "New Dual Descriptor for Chemical Reactivity". The Journal of Physical Chemistry A 109 (1): 205–212. doi:10.1021/jp046577a. PMID 16839107.
- ↑ Hickenboth, Charles R.; Moore, Jeffrey S.; White, Scott R.; Sottos, Nancy R.; Baudry, Jerome; Wilson, Scott R. (2007). "Biasing reaction pathways with mechanical force". Nature 446 (7134): 423–7. doi:10.1038/nature05681. PMID 17377579.
- ↑ Corey, E. J. (2004). "Impossible Dreams". The Journal of Organic Chemistry 69 (9): 2917–9. doi:10.1021/jo049925d. PMID 15104426.
- ↑ Johnson, Carolyn Y. (March 1, 2005) Whose idea was it?. Boston Globe
- ↑ Hoffmann, Roald (2004). "A Claim on the Development of the Frontier Orbital Explanation of Electrocyclic Reactions". Angewandte Chemie International Edition 43 (48): 6586. doi:10.1002/anie.200461440.
- ↑ Corey, E. J.; Hortmann, Alfred G. (1963). "Total Synthesis of Dihydrocostunolide". Journal of the American Chemical Society 85 (24): 4033. doi:10.1021/ja00907a030.
- ↑ Corey, E. J.; Hortmann, Alfred G. (1965). "The total synthesis of dihydrocostunolide". Journal of the American Chemical Society 87 (24): 5736–42. doi:10.1021/ja00952a037. PMID 5845424.