White catalyst
 | |
| Identifiers | |
|---|---|
| 858971-43-4 | |
| Properties | |
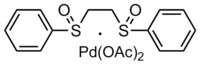
| C18H20O6PdS2 | |
| Molar mass | 502.89 g·mol−1 |
| Appearance | Red-brown solid |
| Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| | |
| Infobox references | |
The White catalyst is a transition metal coordination complex named after the chemist by whom it was first synthesized, M. Christina White, a professor at the University of Illinois. The catalyst has been used in a variety of allylic C-H functionalization reactions of α-olefins. In addition, it has been shown to catalyze oxidative Heck reactions.
Preparation
This compound is commercially available. It may be prepared by oxidation of 1,2-bis(phenylthio)ethane to the sulfoxide, followed by reaction with palladium acetate.[1]
Mechanism of allylic C-H acetoxylation
The reaction mechanism of allylic C-H acetoxylation has been studied.[2] The first step in the catalytic cycle is cleavage of the allylic C-H bond. The sulfoxide ligand is thought to promote this step by generating a highly electrophilic, possibly cationic palladium species in situ. This species coordinates to the alkene and acidifies the adjacent C-H bond, which allows acetate to abstract the proton and forms a π-allyl palladium complex (II). Subsequently, a π-acid such as benzoquinone coordinates to the palladium, activating the π-allyl complex to nucleophilic attack (III). A nucleophile, in this case acetate, attacks to reductively eliminate palladium, generating the product and palladium(0) (IV). The palladium(0) is reoxidized to palladium(II) by benzoquinone and the sulfoxide ligand reassociates, closing the catalytic cycle.

Allylic esterification
The White catalyst was originally developed for use in a branched allylic acetoxylation reaction.[2] An enantioselective version of this reaction was subsequently reported, using chromium(III) salen fluoride as a chiral cocatalyst.[3] A macrolactonization reaction based on the branched allylic esterification was developed for the preparation of 14- to 19-membered macrolides.[4] This method was applied to the total synthesis of 6-deoxyerythronolide B.[5] In addition to acetate, a wide variety of carboxylic acids may be employed as nucleophiles in the branch allylic esterification reaction. As the first step in an esterification/Heck sequence, aliphatic and aromatic carboxylates were demonstrated, including amino acids.[6]

Allylic amination
The White catalyst can effect both branched and linear regioselective allylic C-H aminations. In order to promote nucleophilic attack at the internal terminus of the π-allyl to generate branched product, a tethered N-sulfonyl carbamate nucleophile is used. This strategy has been applied to the synthesis of 1,2 and 1,3-amino alcohols.[7][8] The amination proceeds with high yields and good diastereoselectivity, and the products may be readily elaborated to amino acids and other synthetic intermediates and natural products. Key to the development of the reaction was identification of a very acidic nitrogen nucleophile with a pKa close to acetic acid, as more basic nucleophiles divert reactivity to aminopalladation. The intermolecular version of the allylic C-H amination is also known.[9] Using methyl N-tosyl carbamate nucleophile, the linear E-allylic amine products are obtained from α-olefin substrates. It has been shown that functionalization of the π-allyl intermediate may be promoted by chromium(III) salen chloride activation of the electrophile, or Hunig's base activation of the nucleophile.[10]

Allylic alkylation
In 2008, simultaneous publications described the allylic C-H alkylation of allylarene substrates.[11][12] These reactions were catalyzed by the White catalyst or by an earlier version of the complex bearing benzyl substituents on the sulfoxide in place of phenyl. It was demonstrated that an additional sulfoxide ligand, dimethylsulfoxide (DMSO), was essential for promoting functionalization of the π-allyl intermediate; the bis-sulfoxide ligand alone was unable to complete the catalytic cycle.

Heck reaction
The White catalyst has been found to be an effective catalyst for an oxidative version of the classic Heck reaction. Rather than performing allylic C-H cleavage—a relatively slow process—the catalyst quickly transmetallates with a boronic acid. This aryl palladium intermediate undergoes a 1,2-addition across the alkene double bond. β-Hydride elimination releases the product. The oxidative Heck was originally reported as a sequential process following allylic C-H esterification.[6] It was subsequently demonstrated as a stand-alone method for a broad range of α-olefin substrates.[13] The regioselectivity of the reaction is controlled by directing groups such as carbonyls, alcohols and amines.

References
- ↑ Kenneth J. Fraunhoffer and M. Christina White (2007). "syn-1,2-Amino Alcohols via Diastereoselective Allylic C-H Amination". J. Am. Chem. Soc. 129 (23): 7274–7276. doi:10.1021/ja071905g.
- 1 2 Chen, M. S.; Prabagaran, N.; Labenz, N. A.; White, M. C. (2005). "Serial Ligand Catalysis: A Highly Selective Allylic C-H Oxidation". J. Am. Chem. Soc. 127 (19): 6970–6971. doi:10.1021/ja0500198.
- ↑ Covell, D. J.; White, M. C. (2008). "A Chiral Lewis Acid Strategy for Enantioselective Allylic C-H Oxidation". Angewandte Chemie International Edition 47 (34): 6448–6451. doi:10.1002/anie.200802106.
- ↑ Fraunhoffer, K. J.; Prabagaran, N.; Sirois, L. E.; White, M. C. (2006). "Macrolactonization via Hydrocarbon Oxidation". J. Am. Chem. Soc. 128 (28): 9032–9033. doi:10.1021/ja063096r.
- ↑ Stang, E. M.; White, M. C. (2009). "Total synthesis and study of 6-deoxyerythronolide B by late-stage C–H oxidation". Nature Chemistry 1 (7): 547–551. doi:10.1038/NCHEM.351.
- 1 2 Delcamp, J. H.; White, M. C. (2006). "Sequential Hydrocarbon Functionalization: Allylic C-H Oxidation/Vinylic C-H Arylation". J. Am. Chem. Soc. 128 (47): 15076–15077. doi:10.1021/ja066563d.
- ↑ Fraunhoffer, K. J.; White, M. C. (2007). "syn-1,2-Amino Alcohols via Diastereoselective Allylic C-H Amination". J. Am. Chem. Soc. 129 (23): 7274–7276. doi:10.1021/ja071905g.
- ↑ Rice, G. T.; White, M. C. (2009). "Allylic C-H Amination for the Preparation of syn-1,3-Amino Alcohol Motifs". J. Am. Chem. Soc. 131 (33): 11707–11711. doi:10.1021/ja9054959.
- ↑ Reed, S. A.; White, M. C. (2008). "Catalytic Intermolecular Linear Allylic C-H Amination via Heterobimetallic Catalysis". J. Am. Chem. Soc. 130 (11): 3316–3318. doi:10.1021/ja710206u.
- ↑ Reed, S. A.; Mazzotti, A. R.; White, M. C. (2009). "A Catalytic, Brønsted Base Strategy for Intermolecular Allylic C-H Amination". J. Am. Chem. Soc. 131 (33): 11701–11706. doi:10.1021/ja903939k.
- ↑ Young, A. J.; White, M. C. (2008). "Catalytic Intermolecular Allylic C-H Alkylation". J. Am. Chem. Soc. 130 (43): 14090–14091. doi:10.1021/ja806867p.
- ↑ Lin, S.; Song, C.-X.; Cai, G.-X.; Wang, W.-H.; Shi, Z.-J. (2008). "Intra/Intermolecular Direct Allylic Alkylation via Pd(II)-Catalyzed Allylic C-H Activation". J. Am. Chem. Soc. 130 (39): 12901–12903. doi:10.1021/ja803452p.
- ↑ Delcamp, J. H.; Brucks, A. P.; White, M. C. (2008). "A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction". J. Am. Chem. Soc. 130 (34): 11270–11271. doi:10.1021/ja804120r.