Equation of state
In physics and thermodynamics, an equation of state is a relation between state variables.[1] More specifically, an equation of state is a thermodynamic equation describing the state of matter under a given set of physical conditions. It is a constitutive equation which provides a mathematical relationship between two or more state functions associated with the matter, such as its temperature, pressure, volume, or internal energy. Equations of state are useful in describing the properties of fluids, mixtures of fluids, solids, and even the interior of stars.
| Thermodynamics | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
 The classical Carnot heat engine | ||||||||||||
|
Branches |
||||||||||||
|
||||||||||||
| Book:Thermodynamics | ||||||||||||
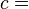

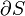
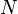
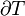
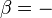
Overview
The most prominent use of an equation of state is to correlate densities of gases and liquids to temperatures and pressures. One of the simplest equations of state for this purpose is the ideal gas law, which is roughly accurate for weakly polar gases at low pressures and moderate temperatures. However, this equation becomes increasingly inaccurate at higher pressures and lower temperatures, and fails to predict condensation from a gas to a liquid. Therefore, a number of more accurate equations of state have been developed for gases and liquids. At present, there is no single equation of state that accurately predicts the properties of all substances under all conditions.
Measurements of equation-of-state parameters, especially at high pressures, can be made using lasers.[2][3][4]
In addition, there are also equations of state describing solids, including the transition of solids from one crystalline state to another. There are equations that model the interior of stars, including neutron stars, dense matter (quark–gluon plasmas) and radiation fields. A related concept is the perfect fluid equation of state used in cosmology.
In practical context, the equations of state are instrumental for PVT calculation in process engineering problems and especially in petroleum gas/liquid equilibrium calculations. A successful PVT model based on a fitting equation of state can be helpful to determine the state of the flow regime, the parameters for handling the reservoir fluids, piping and sizing.
Historical
Boyle's law (1662)
Boyle's Law was perhaps the first expression of an equation of state. In 1662, the noted Irish physicist and chemist Robert Boyle performed a series of experiments employing a J-shaped glass tube, which was sealed on one end. Mercury was added to the tube, trapping a fixed quantity of air in the short, sealed end of the tube. Then the volume of gas was carefully measured as additional mercury was added to the tube. The pressure of the gas could be determined by the difference between the mercury level in the short end of the tube and that in the long, open end. Through these experiments, Boyle noted that the gas volume varied inversely with the pressure. In mathematical form, this can be stated as:
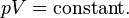
The above relationship has also been attributed to Edme Mariotte and is sometimes referred to as Mariotte's law. However, Mariotte's work was not published until 1676.
Charles's law or Law of Charles and Gay-Lussac (1787)
In 1787 the French physicist Jacques Charles found that oxygen, nitrogen, hydrogen, carbon dioxide, and air expand to the same extent over the same 80 kelvin interval. Later, in 1802, Joseph Louis Gay-Lussac published results of similar experiments, indicating a linear relationship between volume and temperature:
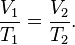
Dalton's law of partial pressures (1801)
Dalton's Law of partial pressure states that the pressure of a mixture of gases is equal to the sum of the pressures of all of the constituent gases alone.
Mathematically, this can be represented for n species as:
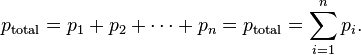
The ideal gas law (1834)
In 1834 Émile Clapeyron combined Boyle's Law and Charles' law into the first statement of the ideal gas law. Initially the law was formulated as pVm = R(TC + 267) (with temperature expressed in degrees Celsius), where R is the gas constant. However, later work revealed that the number should actually be closer to 273.2, and then the Celsius scale was defined with 0 °C = 273.15 K, giving:
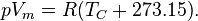
Van der Waals equation of state (1873)
In 1873, J. D. van der Waals introduced the first equation of state derived by the assumption of a finite volume occupied by the constituent molecules.[5] His new formula revolutionized the study of equations of state, and was most famously continued via the Redlich–Kwong equation of state and the Soave modification of Redlich-Kwong.
Major equations of state
For a given amount of substance contained in a system, the temperature, volume, and pressure are not independent quantities; they are connected by a relationship of the general form:
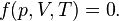
In the following equations the variables are defined as follows. Any consistent set of units may be used, although SI units are preferred. Absolute temperature refers to use of the Kelvin (K) or Rankine (°R) temperature scales, with zero being absolute zero.
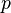 = pressure (absolute)
= pressure (absolute)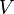 = volume
= volume = number of moles of a substance
= number of moles of a substance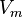 =
= 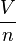 = molar volume, the volume of 1 mole of gas or liquid
= molar volume, the volume of 1 mole of gas or liquid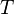 = absolute temperature
= absolute temperature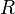 = ideal gas constant (8.3144621 J/(mol·K))
= ideal gas constant (8.3144621 J/(mol·K))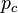 = pressure at the critical point
= pressure at the critical point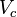 = molar volume at the critical point
= molar volume at the critical point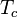 = absolute temperature at the critical point
= absolute temperature at the critical point
Classical ideal gas law
The classical ideal gas law may be written:
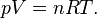
In the form shown above, the equation of state is thus
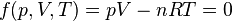 .
.
The ideal gas law may also be expressed as follows
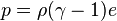
where  is the density,
is the density, 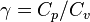 is the adiabatic index (ratio of specific heats),
is the adiabatic index (ratio of specific heats), 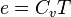 is the internal energy per unit mass (the "specific internal energy"),
is the internal energy per unit mass (the "specific internal energy"), 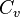 is the specific heat at constant volume, and
is the specific heat at constant volume, and  is the specific heat at constant pressure.
is the specific heat at constant pressure.
Cubic equations of state
Cubic equations of state are called such because they can be rewritten as a cubic function of Vm.
Van der Waals equation of state
The Van der Waals equation of state may be written:
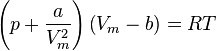
where  is molar volume. The substance-specific constants
is molar volume. The substance-specific constants 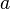 and
and 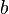 can be calculated from the critical properties
can be calculated from the critical properties 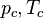 and
and 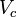 (noting that is the molar volume at the critical point) as:
(noting that is the molar volume at the critical point) as:
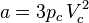
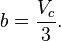
Also written as
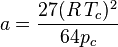
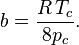
Proposed in 1873, the van der Waals equation of state was one of the first to perform markedly better than the ideal gas law. In this landmark equation is called the attraction parameter and the repulsion parameter or the effective molecular volume. While the equation is definitely superior to the ideal gas law and does predict the formation of a liquid phase, the agreement with experimental data is limited for conditions where the liquid forms. While the van der Waals equation is commonly referenced in text-books and papers for historical reasons, it is now obsolete. Other modern equations of only slightly greater complexity are much more accurate.
The van der Waals equation may be considered as the ideal gas law, "improved" due to two independent reasons:
- Molecules are thought as particles with volume, not material points. Thus cannot be too little, less than some constant. So we get (
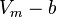 ) instead of .
) instead of . - While ideal gas molecules do not interact, we consider molecules attracting others within a distance of several molecules' radii. It makes no effect inside the material, but surface molecules are attracted into the material from the surface. We see this as diminishing of pressure on the outer shell (which is used in the ideal gas law), so we write (
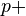 something) instead of
something) instead of 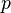 . To evaluate this ‘something’, let's examine an additional force acting on an element of gas surface. While the force acting on each surface molecule is ~, the force acting on the whole element is ~
. To evaluate this ‘something’, let's examine an additional force acting on an element of gas surface. While the force acting on each surface molecule is ~, the force acting on the whole element is ~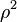 ~
~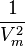 .
.
With the reduced state variables, i.e. Vr=Vm/Vc, Pr=P/Pc and Tr=T/Tc, the reduced form of the Van der Waals equation can be formulated:
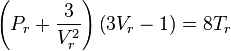
The benefit of this form is that for given Tr and Pr, the reduced volume of the liquid and gas can be calculated directly using Cardano's method for the reduced cubic form:
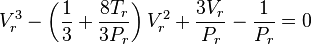
For Pr<1 and Tr<1, the system is in a state of vapor–liquid equilibrium. The reduced cubic equation of state yields in that case 3 solutions. The largest and the lowest solution are the gas and liquid reduced volume.
Redlich-Kwong equation of state
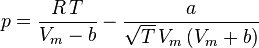
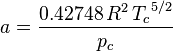
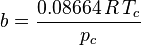
Introduced in 1949, the Redlich-Kwong equation of state was a considerable improvement over other equations of the time. It is still of interest primarily due to its relatively simple form. While superior to the van der Waals equation of state, it performs poorly with respect to the liquid phase and thus cannot be used for accurately calculating vapor–liquid equilibria. However, it can be used in conjunction with separate liquid-phase correlations for this purpose.
The Redlich-Kwong equation is adequate for calculation of gas phase properties when the ratio of the pressure to the critical pressure (reduced pressure) is less than about one-half of the ratio of the temperature to the critical temperature (reduced temperature):

Soave modification of Redlich-Kwong
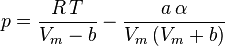
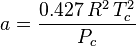
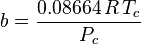
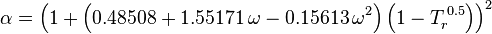
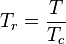
Where ω is the acentric factor for the species.
This formulation for  is due to Graboski and Daubert. The original formulation from Soave is:
is due to Graboski and Daubert. The original formulation from Soave is:
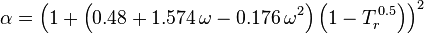
for hydrogen:
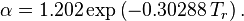
We can also write it in the polynomial form, with:
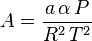
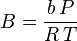
then we have:
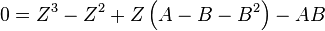
where 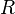 is the universal gas constant and Z=PV/(RT) is the compressibility factor.
is the universal gas constant and Z=PV/(RT) is the compressibility factor.
In 1972 G. Soave[6] replaced the 1/√(T) term of the Redlich-Kwong equation with a function α(T,ω) involving the temperature and the acentric factor (the resulting equation is also known as the Soave-Redlich-Kwong equation). The α function was devised to fit the vapor pressure data of hydrocarbons and the equation does fairly well for these materials.
Note especially that this replacement changes the definition of a slightly, as the  is now to the second power.
is now to the second power.
Peng–Robinson equation of state
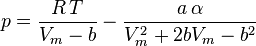
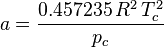

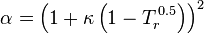
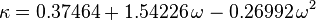
In polynomial form:
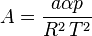
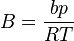
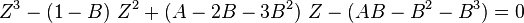
where 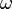 is the acentric factor of the species, is the universal gas constant and Z=PV/(RT) is compressibility factor.
is the acentric factor of the species, is the universal gas constant and Z=PV/(RT) is compressibility factor.
The Peng–Robinson equation was developed in 1976 at The University of Alberta in order to satisfy the following goals:[7]
- The parameters should be expressible in terms of the critical properties and the acentric factor.
- The model should provide reasonable accuracy near the critical point, particularly for calculations of the compressibility factor and liquid density.
- The mixing rules should not employ more than a single binary interaction parameter, which should be independent of temperature pressure and composition.
- The equation should be applicable to all calculations of all fluid properties in natural gas processes.
For the most part the Peng–Robinson equation exhibits performance similar to the Soave equation, although it is generally superior in predicting the liquid densities of many materials, especially nonpolar ones. The departure functions of the Peng–Robinson equation are given on a separate article.
Peng–Robinson-Stryjek-Vera equations of state
PRSV1
A modification to the attraction term in the Peng–Robinson equation of state published by Stryjek and Vera in 1986 (PRSV) significantly improved the model's accuracy by introducing an adjustable pure component parameter and by modifying the polynomial fit of the acentric factor.[8]
The modification is:
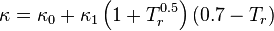
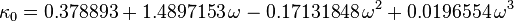
where 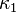 is an adjustable pure component parameter. Stryjek and Vera published pure component parameters for many compounds of industrial interest in their original journal article. At reduced temperatures above 0.7, they recommend to set
is an adjustable pure component parameter. Stryjek and Vera published pure component parameters for many compounds of industrial interest in their original journal article. At reduced temperatures above 0.7, they recommend to set 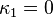 and simply use
and simply use 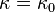 . For alcohols and water the value of may be used up to the critical temperature and set to zero at higher temperatures.[8]
. For alcohols and water the value of may be used up to the critical temperature and set to zero at higher temperatures.[8]
PRSV2
A subsequent modification published in 1986 (PRSV2) further improved the model's accuracy by introducing two additional pure component parameters to the previous attraction term modification.[9]
The modification is:
![\kappa = \kappa_0 + \left[\kappa_1+\kappa_2\left(\kappa_3-T_r\right)\left(1-T_r^{0.5}\right)\right]\left(1+T_r^{0.5}\right) \left(0.7-T_r\right)](../I/m/d55a9f0c6453e1ccf7aa996ce6adc44c.png)
where , 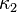 , and
, and 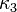 are adjustable pure component parameters.
are adjustable pure component parameters.
PRSV2 is particularly advantageous for VLE calculations. While PRSV1 does offer an advantage over the Peng–Robinson model for describing thermodynamic behavior, it is still not accurate enough, in general, for phase equilibrium calculations.[8] The highly non-linear behavior of phase-equilibrium calculation methods tends to amplify what would otherwise be acceptably small errors. It is therefore recommended that PRSV2 be used for equilibrium calculations when applying these models to a design. However, once the equilibrium state has been determined, the phase specific thermodynamic values at equilibrium may be determined by one of several simpler models with a reasonable degree of accuracy.[9]
One thing to note is that in the PRSV equation, the parameter fit is done in a particular temperature range which is usually below the critical temperature. Above the critical temperature, the PRSV alpha function tends to diverge and become arbitrarily large instead of tending towards 0. Because of this, alternate equations for alpha should be employed above the critical point. This is especially important for systems containing hydrogen which is often found at temperatures far above its critical point. Several alternate formulations have been proposed. Some well known ones are by Twu et all or by Mathias and Copeman.
Elliott, Suresh, Donohue equation of state
The Elliott, Suresh, and Donohue (ESD) equation of state was proposed in 1990.[10] The equation seeks to correct a shortcoming in the Peng–Robinson EOS in that there was an inaccuracy in the van der Waals repulsive term. The EOS accounts for the effect of the shape of a non-polar molecule and can be extended to polymers with the addition of an extra term (not shown). The EOS itself was developed through modeling computer simulations and should capture the essential physics of the size, shape, and hydrogen bonding.
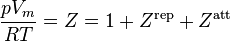
where:
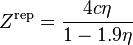
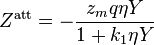
and
 is a "shape factor", with
is a "shape factor", with 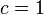 for spherical molecules
for spherical molecules- For non-spherical molecules, the following relation is suggested:
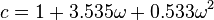 where is the acentric factor
where is the acentric factor
- The reduced number density
 is defined as
is defined as 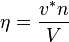
where
 is the characteristic size parameter
is the characteristic size parameter is the number of molecules
is the number of molecules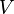 is the volume of the container
is the volume of the container
The characteristic size parameter is related to the shape parameter through
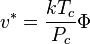
where
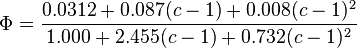 and
and  is Boltzmann's constant.
is Boltzmann's constant.
Noting the relationships between Boltzmann's constant and the Universal gas constant, and observing that the number of molecules can be expressed in terms of Avogadro's number and the molar mass, the reduced number density can be expressed in terms of the molar volume as
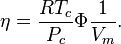
The shape parameter 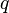 appearing in the Attraction term and the term
appearing in the Attraction term and the term 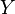 are given by
are given by
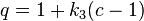 (and is hence also equal to 1 for spherical molecules).
(and is hence also equal to 1 for spherical molecules).
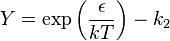
where  is the depth of the square-well potential and is given by
is the depth of the square-well potential and is given by
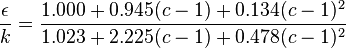
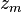 ,
,  ,
,  and
and 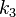 are constants in the equation of state:
are constants in the equation of state: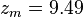 for spherical molecules (c=1)
for spherical molecules (c=1)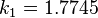 for spherical molecules (c=1)
for spherical molecules (c=1)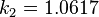 for spherical molecules (c=1)
for spherical molecules (c=1)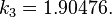
The model can be extended to associating components and mixtures of nonassociating components. Details are in the paper by J.R. Elliott, Jr. et al. (1990).[10]
Non-cubic equations of state
Dieterici equation of state
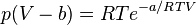
where a is associated with the interaction between molecules and b takes into account the finite size of the molecules, similar to the Van der Waals equation.
The reduced coordinates are:
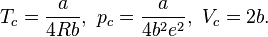
Virial equations of state
Virial equation of state
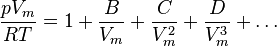
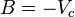
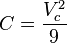
Although usually not the most convenient equation of state, the virial equation is important because it can be derived directly from statistical mechanics. This equation is also called the Kamerlingh Onnes equation. If appropriate assumptions are made about the mathematical form of intermolecular forces, theoretical expressions can be developed for each of the coefficients. In this case B corresponds to interactions between pairs of molecules, C to triplets, and so on. Accuracy can be increased indefinitely by considering higher order terms. The coefficients B, C, D, etc. are functions of temperature only.
It can also be used to work out the Boyle Temperature (the temperature at which B = 0 and ideal gas laws apply) from a and b from the Van der Waals equation of state, if you use the value for B shown below:
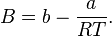
The BWR equation of state
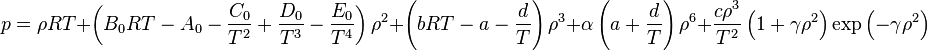
where
- p = pressure
- ρ = the molar density
Values of the various parameters for 15 substances can be found in K.E. Starling (1973). Fluid Properties for Light Petroleum Systems. Gulf Publishing Company.
Multiparameter equations of state
Helmholtz Function form
Multiparameter equations of state (MEOS) can be used to represent pure fluids with high accuracy, in both the liquid and gaseous states. MEOS's represent the Helmholtz function of the fluid as the sum of ideal gas and residual terms. Both terms are explicit in reduced temperature and reduced density - thus:
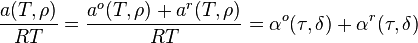
Where:
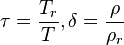
The reduced density and temperature are typically, though not always, the critical values for the pure fluid. Other thermodynamic functions can be derived from the MEOS by using appropriate derivatives of the Helmholtz function; hence, because integration of the MEOS is not required, there are few restrictions as to the functional form of the ideal or residual terms. Typical MEOS use upwards of 50 fluid specific parameters, but are able to represent the fluid's properties with high accuracy. MEOS are available currently for about 50 of the most common industrial fluids including refrigerants. Mixture models also exist.
Other equations of state of interest
Stiffened equation of state
When considering water under very high pressures (typical applications are underwater nuclear explosions, sonic shock lithotripsy, and sonoluminescence) the stiffened equation of state is often used:
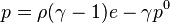
where  is the internal energy per unit mass,
is the internal energy per unit mass, 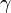 is an empirically determined constant typically taken to be about 6.1, and
is an empirically determined constant typically taken to be about 6.1, and 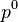 is another constant, representing the molecular attraction between water molecules. The magnitude of the correction is about 2 gigapascals (20,000 atmospheres).
is another constant, representing the molecular attraction between water molecules. The magnitude of the correction is about 2 gigapascals (20,000 atmospheres).
The equation is stated in this form because the speed of sound in water is given by 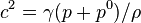 .
.
Thus water behaves as though it is an ideal gas that is already under about 20,000 atmospheres (2 GPa) pressure, and explains why water is commonly assumed to be incompressible: when the external pressure changes from 1 atmosphere to 2 atmospheres (100 kPa to 200 kPa), the water behaves as an ideal gas would when changing from 20,001 to 20,002 atmospheres (2000.1 MPa to 2000.2 MPa).
This equation mispredicts the specific heat capacity of water but few simple alternatives are available for severely nonisentropic processes such as strong shocks.
Ultrarelativistic equation of state
An ultrarelativistic fluid has equation of state
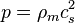
where is the pressure,  is the mass density, and
is the mass density, and  is the speed of sound.
is the speed of sound.
Ideal Bose equation of state
The equation of state for an ideal Bose gas is
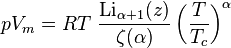
where α is an exponent specific to the system (e.g. in the absence of a potential field, α=3/2), z is exp(μ/kT) where μ is the chemical potential, Li is the polylogarithm, ζ is the Riemann zeta function, and Tc is the critical temperature at which a Bose–Einstein condensate begins to form.
Jones-Wilkins-Lee equation of state for explosives (JWL-equation)
The equation of state from Jones-Wilkins-Lee is used to describe the detonation products of explosives.
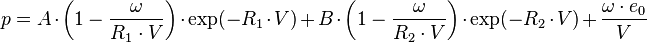
The ratio 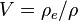 is defined by using
is defined by using 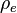 = density of the explosive (solid part) and = density of the detonation products. The parameters
= density of the explosive (solid part) and = density of the detonation products. The parameters 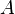 ,
, 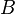 ,
,  ,
, 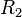 and are given by several references.[11] In addition, the initial density (solid part)
and are given by several references.[11] In addition, the initial density (solid part) 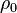 , speed of detonation
, speed of detonation 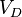 , Chapman–Jouguet pressure
, Chapman–Jouguet pressure 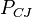 and the chemical energy of the explosive
and the chemical energy of the explosive 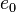 are given in such references. These parameters are obtained by fitting the JWL-EOS to experimental results. Typical parameters for some explosives are listed in the table below.
are given in such references. These parameters are obtained by fitting the JWL-EOS to experimental results. Typical parameters for some explosives are listed in the table below.
| Material | 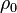 (g/cm3) (g/cm3) |
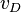 (m/s) (m/s) |
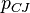 (GPa) (GPa) |
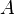 (GPa) (GPa) |
 (GPa) (GPa) |
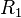 |
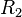 |
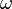 |
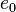 (GPa) (GPa) |
|---|---|---|---|---|---|---|---|---|---|
| TNT | 1.630 | 6930 | 21.0 | 373.8 | 3.747 | 4.15 | 0.90 | 0.35 | 6.00 |
| Composition B | 1.717 | 7980 | 29.5 | 524.2 | 7.678 | 4.20 | 1.10 | 0.35 | 8.50 |
| PBX 9501[12] | 1.844 | 36.3 | 852.4 | 18.02 | 4.55 | 1.3 | 0.38 | 10.2 |
Equations of state for solids and liquids
Common abbreviations: 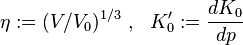
- Tait equation for water and other liquids. Several equations are referred to as the Tait equation.
- Murnaghan equation of state
![p(V) = \frac{K_0}{K_0'} \left[\eta^{-3K_0'} - 1\right]](../I/m/d42178d21fde72ee6675704a4a7e021c.png)
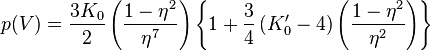
- Stacey-Brennan-Irvine equation of state[13] (falsely often refer to Rose-Vinet equation of state
![p(V)=3K_0\left(\frac{1-\eta}{\eta^2}\right)\exp\left[\tfrac{3}{2}\left(K_0^\prime-1\right)(1-\eta)\right]](../I/m/268a483868b0d8881ce490427fb3d7ba.png)
![p(V)=3K_0\left(\frac{1-\eta}{\eta^5}\right)\exp\left[\tfrac{3}{2}\left(K_0^\prime-3\right)(1-\eta)\right]](../I/m/8b3dd5a7bf06eb48678b5655688b4439.png)
- Adapted Polynomial equation of state[17] ( second order form = AP2, adapted for extreme compression)
![p(V)=3K_0\left(\frac{1-\eta}{\eta^5}\right)\exp\left[c_0(1-\eta)\right]\left\{1+c_2\eta(1-\eta)\right\}](../I/m/bc8446dc19d8570d62ec93c1a58d9d3e.png)
- with
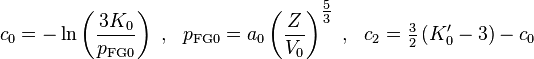
- where
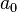 = 0.02337 GPa.nm5. The total number of electrons
= 0.02337 GPa.nm5. The total number of electrons 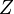 in the initial volume
in the initial volume 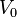 determines the Fermi gas pressure
determines the Fermi gas pressure 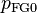 , which provides for the correct behavior at extreme compression. So far there are no known "simple" solids that require higher order terms.
, which provides for the correct behavior at extreme compression. So far there are no known "simple" solids that require higher order terms.
- Adapted polynomial equation of state[17] (third order form = AP3)
![p(V)=3K_0\left(\frac{1-\eta}{\eta^5}\right)\exp\left[c_0(1-\eta)\right]\left\{1+c_2\eta(1-\eta)+c_3\eta(1-\eta)^2\right\}](../I/m/5cbe4b84c1a02322935c1b738e9971f0.png)
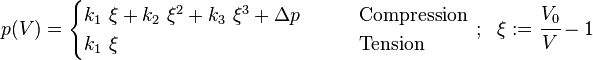
- Mie–Gruneisen equation of state (for a more detailed discussion see ref.[18])
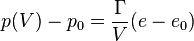
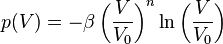
- where
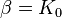 is the bulk modulus at equilibrium volume and
is the bulk modulus at equilibrium volume and 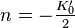 typically about -2 is often related to the Grüneisen parameter by
typically about -2 is often related to the Grüneisen parameter by 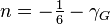
See also
References
- ↑ Perrot, Pierre (1998). A to Z of Thermodynamics. Oxford University Press. ISBN 0-19-856552-6.
- ↑ Solem, J. C.; Veeser, L. (1977). "Exploratory laser-driven shock wave studies" (PDF). Los Alamos Scientific Laboratory Report LA-6997.
- ↑ Veeser, L. R.; Solem, J. C. (1978). "Studies of Laser-driven shock waves in aluminum". Physical Review Letters 40 (21): 1391.
- ↑ Veeser, L.; Solem, J. C.; Lieber, A. (1979). "Impedance-match experiments using laser-driven shock waves". Applied Physics Letters 35: 761.
- ↑ van der Waals, J. D. (1873). On the Continuity of the Gaseous and Liquid States (doctoral dissertation). Universiteit Leiden.
- ↑ Soave, G. Equilibrium Constants from a Modified Redlich-Kwong Equation of State, Chem. Eng. Sci.,1 9 7 2, 27, 1197-1203
- ↑ Peng, D. Y., and Robinson, D. B. (1976). "A New Two-Constant Equation of State". Industrial and Engineering Chemistry: Fundamentals 15: 59–64. doi:10.1021/i160057a011.
- 1 2 3 Stryjek, R. and Vera, J. H. (1986). "PRSV: An improved Peng–Robinson equation of state for pure compounds and mixtures". The Canadian Journal of Chemical Engineering 64: 323–333. doi:10.1002/cjce.5450640224.
- 1 2 Stryjek, R. and Vera, J. H. (1986). "PRSV2: A cubic equation of state for accurate vapor—liquid equilibria calculations". The Canadian Journal of Chemical Engineering 64: 820–826. doi:10.1002/cjce.5450640516.
- 1 2 J. Richard, Jr. Elliott, S. Jayaraman Suresh, Marc D. Donohue (1990). "A Simple Equation of State for Nonspherical and Associating Molecules". Ind. Eng. Chem. Res. 29 (7): 1476–1485. doi:10.1021/ie00103a057.
- ↑ B. M. Dobratz, P. C. Crawford (1985). LLNL Explosives Handbook: Properties of Chemical Explosives and Explosive Simulants. University of California; Lawrence Livermore National Laboratory; Report UCRL-5299; Rev.2;.
- ↑ Wilkins, Mark L. (1999), Computer Simulation of Dynamic Phenomena, Springer, p. 80
- ↑ Stacey, F. D.; Brennan, B. J.; Irvine, R. D. (1981). "Finite strain theories and comparisons with seismological data". Surveys in Geophysics 4 (4): 189–232.
- ↑ Holzapfel, W. B. (1991). "Equations of states and scaling rules for molecular solids under strong compression" in "Molecular systems under high pressure" ed. R. Pucci and G. Piccino. North-Holland: Elsevier. pp. 61–68.
- ↑ Holzapfel, W. B. (1991). "Equations of state for solids under strong compression". High Press. Res. 7, 290-293.
- ↑ Holzapfel, Wi. B. (1996). "Physics of solids under strong compression". Rep. Prog. Phys. 59, 29-90.
- 1 2 Holzapfel, W. B. (1998). "Equation of state for solids under strong compression". High Press. Res. 16, 81-126.
- ↑ Holzapfel, W. B. (2004). "Equations of state and thermophysical properties of solids under pressure" in "High-Pressure Crystallography" ed. A. Katrusiak and P. McMillan. Netherlands: Kluver Academic. pp. 217–236.
- Elliot & Lira, (1999). Introductory Chemical Engineering Thermodynamics, Prentice Hall.
| ||||||||||||||||||||||||||||||||||||
