Verlet integration
Verlet integration (French pronunciation: [vɛʁˈlɛ]) is a numerical method used to integrate Newton's equations of motion.[1] It is frequently used to calculate trajectories of particles in molecular dynamics simulations and computer graphics. The algorithm was first used in 1791 by Delambre, and has been rediscovered many times since then, most recently by Loup Verlet in the 1960s for use in molecular dynamics. It was also used by Cowell and Crommelin in 1909 to compute the orbit of Halley's Comet, and by Carl Størmer in 1907 to study the trajectories of electrical particles in a magnetic field (hence it is also called Störmer's method).[2] The Verlet integrator provides good numerical stability, as well as other properties that are important in physical systems such as time-reversibility and preservation of the symplectic form on phase space, at no significant additional computational cost over the simple Euler method.
Basic Störmer–Verlet
For a differential equation of second order of the type 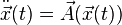 with initial conditions
with initial conditions 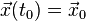 and
and 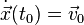 , an approximate numerical solution
, an approximate numerical solution 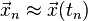 at the times
at the times 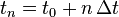 with step size
with step size 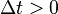 can be obtained by the following method:
can be obtained by the following method:
- set
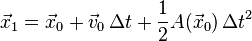
- for n=1,2,... iterate
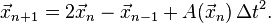
Equations of motion
Newton's equation of motion for conservative physical systems is
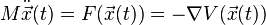
or individually
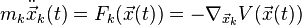
where
- t is the time,
-
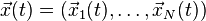 is the ensemble of the position vector of N objects,
is the ensemble of the position vector of N objects, - V is the scalar potential function,
- F is the negative gradient of the potential giving the ensemble of forces on the particles,
- M is the mass matrix, typically diagonal with blocks with mass
 for every particle.
for every particle.
This equation, for various choices of the potential function V, can be used to describe the evolution of diverse physical systems, from the motion of interacting molecules to the orbit of the planets.
After a transformation to bring the mass to the right side and forgetting the structure of multiple particles, the equation may be simplified to
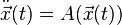
with some suitable vector valued function A representing the position dependent acceleration. Typically, an initial position 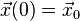 and an initial velocity
and an initial velocity 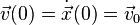 are also given.
are also given.
Verlet integration (without velocities)
To discretize and numerically solve this initial value problem, a time step is chosen and the sampling point sequence 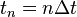 considered. The task is to construct a sequence of points
considered. The task is to construct a sequence of points 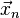 that closely follow the points
that closely follow the points 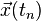 on the trajectory of the exact solution.
on the trajectory of the exact solution.
Where Euler's method uses the forward difference approximation to the first derivative in differential equations of order one, Verlet Integration can be seen as using the central difference approximation to the second derivative:
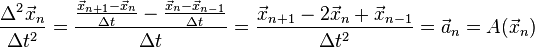
Verlet integration in the form used as the Störmer method[3] uses this equation to obtain the next position vector from the previous two without using the velocity as
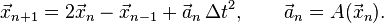
Discretization error
The time symmetry inherent in the method reduces the level of local errors introduced into the integration by the discretization by removing all odd degree terms, here the terms in  of degree three. The local error is quantified by inserting the exact values
of degree three. The local error is quantified by inserting the exact values 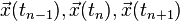 into the iteration and computing the Taylor expansions at time
into the iteration and computing the Taylor expansions at time 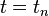 of the position vector
of the position vector 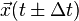 in different time directions.
in different time directions.
![\begin{align}
\vec{x}(t + \Delta t)
&= \vec{x}(t) + \vec{v}(t)\Delta t + \frac{\vec{a}(t) \Delta t^2}{2}
+ \frac{\vec{b}(t) \Delta t^3}{6} + \mathcal{O}(\Delta t^4)\\[0.7em]
\vec{x}(t - \Delta t)
&= \vec{x}(t) - \vec{v}(t)\Delta t + \frac{\vec{a}(t) \Delta t^2}{2}
- \frac{\vec{b}(t) \Delta t^3}{6} + \mathcal{O}(\Delta t^4),\,
\end{align}](../I/m/c3e47660b8558b8ce46541e96f2056be.png)
where  is the position,
is the position, 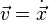 the velocity,
the velocity, 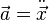 the acceleration and
the acceleration and 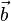 the jerk (third derivative of the position with respect to the time)
the jerk (third derivative of the position with respect to the time)  .
.
Adding these two expansions gives
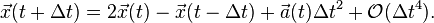
We can see that the first and third-order terms from the Taylor expansion cancel out, thus making the Verlet integrator an order more accurate than integration by simple Taylor expansion alone.
Caution should be applied to the fact that the acceleration here is computed from the exact solution, 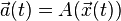 , whereas in the iteration it is computed at the central iteration point,
, whereas in the iteration it is computed at the central iteration point, 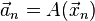 . In computing the global error, that is the distance between exact solution and approximation sequence, those two terms do not cancel exactly, influencing the order of the global error.
. In computing the global error, that is the distance between exact solution and approximation sequence, those two terms do not cancel exactly, influencing the order of the global error.
A simple example
To gain insight into the relation of local and global errors it is helpful to examine simple examples where the exact as well as the approximative solution can be expressed in explicit formulas. The standard example for this task is the exponential function.
Consider the linear differential equation 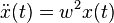 with a constant w. Its exact basis solutions are
with a constant w. Its exact basis solutions are 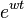 and
and 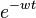 .
.
The Störmer method applied to this differential equation leads to a linear recurrence relation
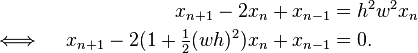
It can be solved by finding the roots of its characteristic polynomial
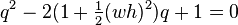 . These are
. These are
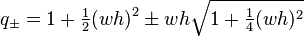 .
.
The basis solutions of the linear recurrence are 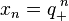 and
and 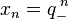 . To compare them with the exact solutions, Taylor expansions are computed.
. To compare them with the exact solutions, Taylor expansions are computed.
![\begin{align}
q_+
&=1+\tfrac12(wh)^2+wh(1+\tfrac18(wh)^2-\tfrac{3}{128}(wh)^4+\mathcal O(h^6))\\[.3em]
&=1+(wh)+\tfrac12(wh)^2+\tfrac18(wh)^3-\tfrac{3}{128}(wh)^5+ \mathcal O(h^7).
\end{align}](../I/m/2436ab20656b9c56070977e39fa590f1.png)
The quotient of this series with the one of the exponential 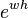 starts with
starts with 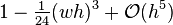 , so
, so
![\begin{align}
q_+&=(1-\tfrac1{24}(wh)^3+\mathcal O(h^5))e^{wh}\\[.3em]
&=e^{-\frac{1}{24}(wh)^3+\mathcal O(h^5)}\,e^{wh}.
\end{align}](../I/m/25ece02a2bc6591bfe37779d28419f7b.png)
From there it follows that for the first basis solution the error can be computed as
![\begin{align}
x_n=q_+^{\;n}&=e^{-\frac{1}{24}(wh)^2\,wt_n+\mathcal O(h^4)}\,e^{wt_n}\\[.3em]
&=e^{wt_n}\left(1-\tfrac{1}{24}(wh)^2\,wt_n+\mathcal O(h^4)\right)\\[.3em]
&=e^{wt_n}+\mathcal O(h^2t_ne^{wt_n}).
\end{align}](../I/m/1e853bcf48312a8303aa868931eaecbe.png)
That is, although the local discretization error is of order 4, due to the second order of the differential equation the global error is of order 2, with a constant that grows exponentially in time.
Starting the iteration
Note that at the start of the Verlet-iteration at step  , time
, time 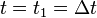 , computing
, computing 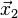 , one already needs the position vector
, one already needs the position vector 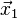 at time
at time 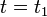 . At first sight this could give problems, because the initial conditions are known only at the initial time
. At first sight this could give problems, because the initial conditions are known only at the initial time 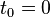 . However, from these the acceleration
. However, from these the acceleration 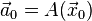 is known, and a suitable approximation for the first time step position can be obtained using the Taylor polynomial of degree two:
is known, and a suitable approximation for the first time step position can be obtained using the Taylor polynomial of degree two:
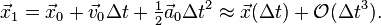
The error on the first time step calculation then is of order 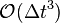 . This is not considered a problem because on a simulation of over a large amount of timesteps, the error on the first timestep is only a negligibly small amount of the total error, which at time
. This is not considered a problem because on a simulation of over a large amount of timesteps, the error on the first timestep is only a negligibly small amount of the total error, which at time  is of the order
is of the order 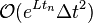 , both for the distance of the position vectors to as for the distance of the divided differences
, both for the distance of the position vectors to as for the distance of the divided differences 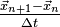 to
to 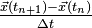 . Moreover, to obtain this second order global error, the initial error needs to be of at least third order.
. Moreover, to obtain this second order global error, the initial error needs to be of at least third order.
Non-constant time differences
A disadvantage of the Störmer–Verlet method is that if the time-step () changes, the method does not approximate the solution to the differential equation. This is particularly an issue for game designers, who may be integrating at a variable framerate.
This can be corrected using the formula:[4]
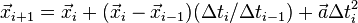
A more exact derivation uses the Taylor series (to second order) at  for times
for times 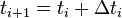 and
and 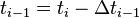 to obtain after elimination of
to obtain after elimination of 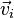
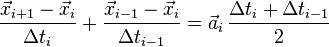
so that the iteration formula becomes
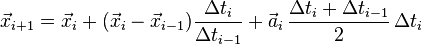
Computing velocities – Störmer–Verlet method
The velocities are not explicitly given in the basic Störmer equation, but often they are necessary for the calculation of certain physical quantities like the kinetic energy. This can create technical challenges in molecular dynamics simulations, because kinetic energy and instantaneous temperatures at time cannot be calculated for a system until the positions are known at time  . This deficiency can either be dealt with using the Velocity Verlet algorithm, or estimating the velocity using the position terms and the mean value theorem:
. This deficiency can either be dealt with using the Velocity Verlet algorithm, or estimating the velocity using the position terms and the mean value theorem:
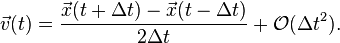
Note that this velocity term is a step behind the position term, since this is for the velocity at time , not , meaning that 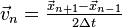 is an order two approximation to
is an order two approximation to 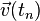 . With the same argument, but halving the time step,
. With the same argument, but halving the time step, 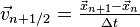 is an order two approximation to
is an order two approximation to 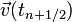 , with
, with 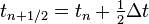 .
.
One can shorten the interval to approximate the velocity at time at the cost of accuracy:
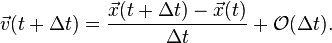
Velocity Verlet
A related, and more commonly used, algorithm is the Velocity Verlet algorithm,[5] similar to the leapfrog method, except that the velocity and position are calculated at the same value of the time variable (Leapfrog does not, as the name suggests). This uses a similar approach but explicitly incorporates velocity, solving the first-timestep problem in the Basic Verlet algorithm:
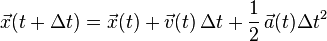
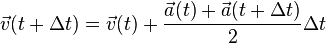
It can be shown that the error on the Velocity Verlet is of the same order as the Basic Verlet. Note that the Velocity algorithm is not necessarily more memory consuming, because it's not necessary to keep track of the velocity at every timestep during the simulation. The standard implementation scheme of this algorithm is:
- Calculate:
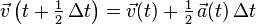
- Calculate:
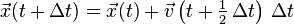
- Derive
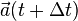 from the interaction potential using
from the interaction potential using 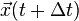
- Calculate:
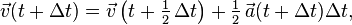
Eliminating the half-step velocity, this algorithm may be shortened to
- Calculate:
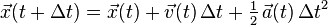
- Derive from the interaction potential using
- Calculate:
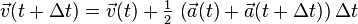
Note, however, that this algorithm assumes that acceleration only depends on position , and does not depend on velocity 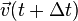 .
.
One might note that the long-term results of Velocity Verlet, and similarly of Leapfrog are one order better than the semi-implicit Euler method. The algorithms are almost identical up to a shifted by half of a timestep in the velocity. This is easily proven by rotating the above loop to start at Step 3 and then noticing that the acceleration term in Step 1 could be eliminated by combining Steps 2 and 4. The only difference is that the midpoint velocity in velocity Verlet is considered the final velocity in semi-implicit Euler method.
The global error of all Euler methods is of order one, whereas the global error of this method is, similar to the midpoint method, of order two. Additionally, if the acceleration indeed results from the forces in a conservative mechanical or Hamiltonian system, the energy of the approximation essentially oscillates around the constant energy of the exactly solved system, with a global error bound again of order one for semi-explicit Euler and order two for Verlet-leapfrog. The same goes for all other conservered quantities of the system like linear or angular momentum, that are always preserved or nearly preserved in a symplectic integrator.[6]
The Velocity Verlet method is a special case of the Newmark-beta method with  and
and 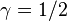 .
.
Error terms
The local error in position of the Verlet integrator is 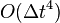 as described above, and the local error in velocity is
as described above, and the local error in velocity is 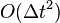 .
.
The global error in position, in contrast, is and the global error in velocity is . These can be derived by noting the following:
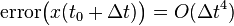
and

Therefore:
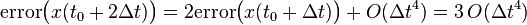
Similarly:
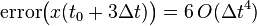
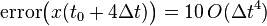
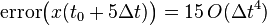
Which can be generalized to (it can be shown by induction, but it is given here without proof):
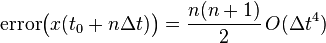
If we consider the global error in position between  and
and 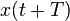 , where
, where 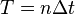 , it is clear that:
, it is clear that:
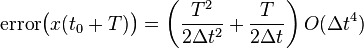
And therefore, the global (cumulative) error over a constant interval of time is given by:
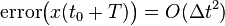
Because the velocity is determined in a non-cumulative way from the positions in the Verlet integrator, the global error in velocity is also .
In molecular dynamics simulations, the global error is typically far more important than the local error, and the Verlet integrator is therefore known as a second-order integrator.
Constraints
Systems of multiple particles with constraints are simpler to solve with Verlet integration than with Euler methods. Constraints between points may be for example potentials constraining them to a specific distance or attractive forces. They may be modeled as springs connecting the particles. Using springs of infinite stiffness, the model may then be solved with a Verlet algorithm.
In one dimension, the relationship between the unconstrained positions 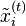 and the actual positions
and the actual positions 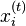 of points i at time t can be found with the algorithm
of points i at time t can be found with the algorithm
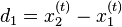
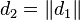
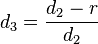
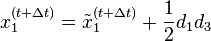
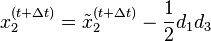
Verlet integration is useful because it directly relates the force to the position, rather than solving the problem via velocities.
Problems, however, arise when multiple constraining forces act on each particle. One way to solve this is to loop through every point in a simulation, so that at every point the constraint relaxation of the last is already used to speed up the spread of the information. In a simulation this may be implemented by using small time steps for the simulation, using a fixed number of constraint solving steps per time step, or solving constraints until they are met by a specific deviation.
When approximating the constraints locally to first order this is the same as the Gauss–Seidel method. For small matrices it is known that LU decomposition is faster. Large systems can be divided into clusters (for example: each ragdoll = cluster). Inside clusters the LU method is used, between clusters the Gauss–Seidel method is used. The matrix code can be reused: The dependency of the forces on the positions can be approximated locally to first order, and the Verlet integration can be made more implicit.
Sophisticated software, such as SuperLU exists to solve complex problems using sparse matrices. Specific techniques, such as using (clusters of) matrices, may be used to address the specific problem, such as that of force propagating through a sheet of cloth without forming a sound wave.[7]
Another way to solve holonomic constraints is to use constraint algorithms.
Collision reactions
One way of reacting to collisions is to use a penalty-based system which basically applies a set force to a point upon contact. The problem with this is that it is very difficult to choose the force imparted. Use too strong a force and objects will become unstable, too weak and the objects will penetrate each other. Another way is to use projection collision reactions which takes the offending point and attempts to move it the shortest distance possible to move it out of the other object.
The Verlet integration would automatically handle the velocity imparted via the collision in the latter case, however note that this is not guaranteed to do so in a way that is consistent with collision physics (that is, changes in momentum are not guaranteed to be realistic). Instead of implicitly changing the velocity term, one would need to explicitly control the final velocities of the objects colliding (by changing the recorded position from the previous time step).
The two simplest methods for deciding on a new velocity are perfectly elastic collisions and inelastic collisions. A slightly more complicated strategy that offers more control would involve using the coefficient of restitution.
See also
- Courant–Friedrichs–Lewy condition
- Energy drift
- Symplectic integrator
- Leapfrog integration
- Beeman's algorithm
Literature
- ↑ Verlet, Loup (1967). "Computer "Experiments" on Classical Fluids. I. Thermodynamical Properties of Lennard−Jones Molecules". Physical Review 159: 98–103. doi:10.1103/PhysRev.159.98.
- ↑ Press, WH; Teukolsky, SA; Vetterling, WT; Flannery, BP (2007). "Section 17.4. Second-Order Conservative Equations". Numerical Recipes: The Art of Scientific Computing (3rd ed.). New York: Cambridge University Press. ISBN 978-0-521-88068-8.
- ↑ webpage with a description of the Störmer method
- ↑ Dummer, Jonathan. "A Simple Time-Corrected Verlet Integration Method".
- ↑ Swope, William C.; H.C. Andersen; P.H. Berens; K.R. Wilson (1 January 1982). "A computer simulation method for the calculation of equilibrium constants for the formation of physical clusters of molecules: Application to small water clusters". The Journal of Chemical Physics 76 (1): 648(Appendix). doi:10.1063/1.442716.
- ↑ Hairer, Ernst; Lubich, Christian; Wanner, Gerhard (2003). "Geometric numerical integration illustrated by the Störmer/Verlet method". Acta Numerica 12: 399–450. doi:10.1017/S0962492902000144.
- ↑ Baraff, D.; Witkin, A. (1998). "Large Steps in Cloth Simulation" (PDF). Computer Graphics Proceedings. Annual Conference Series: 43-54.
External links
- Verlet Integration Demo and Code as a Java Applet
- Advanced Character Physics by Thomas Jakobsen
- The Verlet algorithm
- Theory of Molecular Dynamics Simulations – bottom of page
| ||||||||||||||