Angiogenesis
Angiogenesis is the physiological process through which new blood vessels form from pre-existing vessels.[1][2] This is distinct from vasculogenesis, which is the de novo formation of endothelial cells from mesoderm cell precursors.[3] The first vessels in the developing embryo form through vasculogenesis, after which angiogenesis is responsible for most, if not all, blood vessel growth during development and in disease.[4]
Angiogenesis is a normal and vital process in growth and development, as well as in wound healing and in the formation of granulation tissue. However, it is also a fundamental step in the transition of tumors from a benign state to a malignant one, leading to the use of angiogenesis inhibitors in the treatment of cancer. The essential role of angiogenesis in tumor growth was first proposed in 1971 by Judah Folkman, who described tumors as "hot and bloody."[5]
Types
Sprouting angiogenesis
Sprouting angiogenesis was the first identified form of angiogenesis. It occurs in several well-characterized stages. First, biological signals known as angiogenic growth factors activate receptors on endothelial cells present in pre-existing blood vessels. Second, the activated endothelial cells begin to release enzymes called proteases that degrade the basement membrane to allow endothelial cells to escape from the original (parent) vessel walls. The endothelial cells then proliferate into the surrounding matrix and form solid sprouts connecting neighboring vessels. As sprouts extend toward the source of the angiogenic stimulus, endothelial cells migrate in tandem, using adhesion molecules called integrins. These sprouts then form loops to become a full-fledged vessel lumen as cells migrate to the site of angiogenesis. Sprouting occurs at a rate of several millimeters per day, and enables new vessels to grow across gaps in the vasculature. It is markedly different from splitting angiogenesis because it forms entirely new vessels as opposed to splitting existing vessels.
Intussusceptive angiogenesis
By Intussusception, also known as splitting angiogenesis, a new blood vessel is created by splitting of an existing blood vessel in two.
Intussusception was first observed in neonatal rats. In this type of vessel formation, the capillary wall extends into the lumen to split a single vessel in two. There are four phases of intussusceptive angiogenesis. First, the two opposing capillary walls establish a zone of contact. Second, the endothelial cell junctions are reorganized and the vessel bilayer is perforated to allow growth factors and cells to penetrate into the lumen. Third, a core is formed between the 2 new vessels at the zone of contact that is filled with pericytes and myofibroblasts. These cells begin laying collagen fibers into the core to provide an extracellular matrix for growth of the vessel lumen. Finally, the core is fleshed out with no alterations to the basic structure. Intussusception is important because it is a reorganization of existing cells. It allows a vast increase in the number of capillaries without a corresponding increase in the number of endothelial cells. This is especially important in embryonic development as there are not enough resources to create a rich microvasculature with new cells every time a new vessel develops.[6]
Physiology
Mechanical stimulation
Mechanical stimulation of angiogenesis is not well characterized. There is a significant amount of controversy with regard to shear stress acting on capillaries to cause angiogenesis, although current knowledge suggests that increased muscle contractions may increase angiogenesis.[7] This may be due to an increase in the production of nitric oxide during exercise. Nitric oxide results in vasodilation of blood vessels.
Chemical stimulation
Chemical stimulation of angiogenesis is performed by various angiogenic proteins, including several growth factors.
Overview
| Stimulator | Mechanism |
|---|---|
| FGF | Promotes proliferation & differentiation of endothelial cells, smooth muscle cells, and fibroblasts |
| VEGF | Affects permeability |
| VEGFR and NRP-1 | Integrate survival signals |
| Ang1 and Ang2 | Stabilize vessels |
| PDGF (BB-homodimer) and PDGFR | recruit smooth muscle cells |
| TGF-β, endoglin and TGF-β receptors | ↑extracellular matrix production |
| MCP-1 | |
| Histamine | |
| Integrins αVβ3, αVβ5 (?[8]) and α5β1 | Bind matrix macromolecules and proteinases |
| VE-cadherin and CD31 | endothelial junctional molecules |
| ephrin | Determine formation of arteries or veins |
| plasminogen activators | remodels extracellular matrix, releases and activates growth factors |
| plasminogen activator inhibitor-1 | stabilizes nearby vessels |
| eNOS and COX-2 | |
| AC133 | regulates angioblast differentiation |
| ID1/ID3 | Regulates endothelial transdifferentiation |
FGF
The fibroblast growth factor (FGF) family with its prototype members FGF-1 (acidic FGF) and FGF-2 (basic FGF) consists to date of at least 22 known members.[9] Most are single-chain peptides of 16-18 kDa and display high affinity to heparin and heparan sulfate. In general, FGFs stimulate a variety of cellular functions by binding to cell surface FGF-receptors in the presence of heparin proteoglycans. The FGF-receptor family is composed of seven members, and all the receptor proteins are single-chain receptor tyrosine kinases that become activated through autophosphorylation induced by a mechanism of FGF-mediated receptor dimerization. Receptor activation gives rise to a signal transduction cascade that leads to gene activation and diverse biological responses, including cell differentiation, proliferation, and matrix dissolution, thus initiating a process of mitogenic activity critical for the growth of endothelial cells, fibroblasts, and smooth muscle cells. FGF-1, unique among all 22 members of the FGF family, can bind to all seven FGF-receptor subtypes, making it the broadest-acting member of the FGF family, and a potent mitogen for the diverse cell types needed to mount an angiogenic response in damaged (hypoxic) tissues, where upregulation of FGF-receptors occurs.[10] FGF-1 stimulates the proliferation and differentiation of all cell types necessary for building an arterial vessel, including endothelial cells and smooth muscle cells; this fact distinguishes FGF-1 from other pro-angiogenic growth factors, such as vascular endothelial growth factor (VEGF), which primarily drives the formation of new capillaries.[11][12]
Until 2007, three human clinical trials have been successfully completed with FGF-1, in which the angiogenic protein was injected directly into the damaged heart muscle.[13][14][15][16] Also, one additional human FGF-1 trial has been completed to promote wound healing in diabetics with chronic wounds.
Besides FGF-1, one of the most important functions of fibroblast growth factor-2 (FGF-2 or bFGF) is the promotion of endothelial cell proliferation and the physical organization of endothelial cells into tube-like structures, thus promoting angiogenesis. FGF-2 is a more potent angiogenic factor than VEGF or PDGF (platelet-derived growth factor); however, it is less potent than FGF-1. As well as stimulating blood vessel growth, aFGF (FGF-1) and bFGF (FGF-2) are important players in wound healing. They stimulate the proliferation of fibroblasts and endothelial cells that give rise to angiogenesis and developing granulation tissue; both increase blood supply and fill up a wound space/cavity early in the wound-healing process.
VEGF
Vascular endothelial growth factor (VEGF) has been demonstrated to be a major contributor to angiogenesis, increasing the number of capillaries in a given network. Initial in vitro studies demonstrated bovine capillary endothelial cells will proliferate and show signs of tube structures upon stimulation by VEGF and bFGF, although the results were more pronounced with VEGF.[17] Upregulation of VEGF is a major component of the physiological response to exercise and its role in angiogenesis is suspected to be a possible treatment in vascular injuries.[18][19][20][21] In vitro studies clearly demonstrate that VEGF is a potent stimulator of angiogenesis because, in the presence of this growth factor, plated endothelial cells will proliferate and migrate, eventually forming tube structures resembling capillaries.[7] VEGF causes a massive signaling cascade in endothelial cells. Binding to VEGF receptor-2 (VEGFR-2) starts a tyrosine kinase signaling cascade that stimulates the production of factors that variously stimulate vessel permeability (eNOS, producing NO), proliferation/survival (bFGF), migration (ICAMs/VCAMs/MMPs) and finally differentiation into mature blood vessels. Mechanically, VEGF is upregulated with muscle contractions as a result of increased blood flow to affected areas. The increased flow also causes a large increase in the mRNA production of VEGF receptors 1 and 2. The increase in receptor production means muscle contractions could cause upregulation of the signaling cascade relating to angiogenesis. As part of the angiogenic signaling cascade, NO is widely considered to be a major contributor to the angiogenic response because inhibition of NO significantly reduces the effects of angiogenic growth factors. However, inhibition of NO during exercise does not inhibit angiogenesis, indicating there are other factors involved in the angiogenic response.[7]
Angiopoietins
The angiopoietins, Ang1 and Ang2, are required for the formation of mature blood vessels, as demonstrated by mouse knock out studies.[22] Ang1 and Ang2 are protein growth factors which act by binding their receptors, Tie-1 and Tie-2; while this is somewhat controversial, it seems that cell signals are transmitted mostly by Tie-2; though some papers show physiologic signaling via Tie-1 as well. These receptors are tyrosine kinases. Thus, they can initiate cell signaling when ligand binding causes a dimerization that initiates phosphorylation on key tyrosines.
MMP
Another major contributor to angiogenesis is matrix metalloproteinase (MMP). MMPs help degrade the proteins that keep the vessel walls solid. This proteolysis allows the endothelial cells to escape into the interstitial matrix as seen in sprouting angiogenesis. Inhibition of MMPs prevents the formation of new capillaries.[23] These enzymes are highly regulated during the vessel formation process because destruction of the extracellular matrix would decrease the integrity of the microvasculature.[7]
DII4
Delta-like ligand 4 (DII4) is a protein with a negative regulatory effect on angiogenesis.[24] Dll4 is a transmembrane ligand, for the notch family of receptors.
Chemical inhibition
Angiogenesis inhibitor can be endogenous or come from outside as drug or a dietary component.
Application in medicine
Angiogenesis as a therapeutic target
Angiogenesis may be a target for combating diseases characterized by either poor vascularisation or abnormal vasculature.[25] Application of specific compounds that may inhibit or induce the creation of new blood vessels in the body may help combat such diseases. The presence of blood vessels where there should be none may affect the mechanical properties of a tissue, increasing the likelihood of failure. The absence of blood vessels in a repairing or otherwise metabolically active tissue may inhibit repair or other essential functions. Several diseases, such as ischemic chronic wounds, are the result of failure or insufficient blood vessel formation and may be treated by a local expansion of blood vessels, thus bringing new nutrients to the site, facilitating repair. Other diseases, such as age-related macular degeneration, may be created by a local expansion of blood vessels, interfering with normal physiological processes.
The modern clinical application of the principle of angiogenesis can be divided into two main areas: anti-angiogenic therapies, which angiogenic research began with, and pro-angiogenic therapies. Whereas anti-angiogenic therapies are being employed to fight cancer and malignancies,[26][27] which require an abundance of oxygen and nutrients to proliferate, pro-angiogenic therapies are being explored as options to treat cardiovascular diseases, the number one cause of death in the Western world. One of the first applications of pro-angiogenic methods in humans was a German trial using fibroblast growth factor 1 (FGF-1) for the treatment of coronary artery disease.[11][13][28] Clinical research in therapeutic angiogenesis is ongoing for a variety of atherosclerotic diseases, like coronary heart disease, peripheral arterial disease, wound healing disorders, etc.[14]
Also, regarding the mechanism of action, pro-angiogenic methods can be differentiated into three main categories: gene-therapy, targeting genes of interest for amplification or inhibition; protein-therapy, which primarily manipulates angiogenic growth factors like FGF-1 or vascular endothelial growth factor, VEGF; and cell-based therapies, which involve the implantation of specific cell types.
There are still serious, unsolved problems related to gene therapy. Difficulties include effective integration of the therapeutic genes into the genome of target cells, reducing the risk of an undesired immune response, potential toxicity, immunogenicity, inflammatory responses, and oncogenesis related to the viral vectors used in implanting genes and the sheer complexity of the genetic basis of angiogenesis. The most commonly occurring disorders in humans, such as heart disease, high blood pressure, diabetes and Alzheimer's disease, are most likely caused by the combined effects of variations in many genes, and, thus, injecting a single gene may not be significantly beneficial in such diseases.
In contrast, pro-angiogenic protein therapy uses well-defined, precisely structured proteins, with previously defined optimal doses of the individual protein for disease states, and with well-known biological effects. On the other hand, an obstacle of protein therapy is the mode of delivery. Oral, intravenous, intra-arterial, or intramuscular routes of protein administration are not always as effective, as the therapeutic protein may be metabolized or cleared before it can enter the target tissue. Cell-based pro-angiogenic therapies are still early stages of research, with many open questions regarding best cell types and dosages to use.
Tumor angiogenesis

Cancer cells are cells that have lost their ability to divide in a controlled fashion. A malignant tumor consists of a population of rapidly dividing and growing cancer cells that progressively accrues mutations. However, tumors need a dedicated blood supply to provide the oxygen and other essential nutrients they require in order to grow beyond a certain size (generally 1–2 mm3).[29]
Tumors induce blood vessel growth (angiogenesis) by secreting various growth factors (e.g. VEGF). Growth factors such as bFGF and VEGF can induce capillary growth into the tumor, which some researchers suspect supply required nutrients, allowing for tumor expansion. Unlike normal blood vessels, tumor blood vessels are dilated with an irregular shape.[30] In 2007, it was discovered that cancerous cells stop producing the anti-VEGF enzyme PKG. In normal cells (but not in cancerous ones), PKG apparently limits beta-catenin, which solicits angiogenesis.[31] Other clinicians believe angiogenesis really serves as a waste pathway, taking away the biological end products secreted by rapidly dividing cancer cells. In either case, angiogenesis is a necessary and required step for transition from a small harmless cluster of cells, often said to be about the size of the metal ball at the end of a ball-point pen, to a large tumor. Angiogenesis is also required for the spread of a tumor, or metastasis. Single cancer cells can break away from an established solid tumor, enter the blood vessel, and be carried to a distant site, where they can implant and begin the growth of a secondary tumor. Evidence now suggests the blood vessel in a given solid tumor may, in fact, be mosaic vessels, composed of endothelial cells and tumor cells. This mosaicity allows for substantial shedding of tumor cells into the vasculature, possibly contributing to the appearance of circulating tumor cells in the peripheral blood of patients with malignancies.[32] The subsequent growth of such metastases will also require a supply of nutrients and oxygen and a waste disposal pathway.
Endothelial cells have long been considered genetically more stable than cancer cells. This genomic stability confers an advantage to targeting endothelial cells using antiangiogenic therapy, compared to chemotherapy directed at cancer cells, which rapidly mutate and acquire 'drug resistance' to treatment. For this reason, endothelial cells are thought to be an ideal target for therapies directed against them.[33] Recent studies by Klagsbrun, et al. have shown, however, that endothelial cells growing within tumors do carry genetic abnormalities. Thus, tumor vessels have the theoretical potential for developing acquired resistance to drugs. This is a new area of angiogenesis research being actively pursued.
Two independent studies published in the journal Nature[34][35] in 2010 November confirmed the ability of tumors to make their own blood vessels. When one group found that tumor stem cells could make their own blood vessels and avastin could not inhibit their early differentiation, the other group showed that selective targeting of endothelial cells generated by tumor-derived stem cells in mouse xenografts resulted in tumour reduction.[36] These studies done in glioblastoma model may have implications in other tumors.
Formation of tumor blood vessels
Angiogenesis research is a cutting-edge field in cancer research, and recent evidence also suggests traditional therapies, such as radiation therapy, may actually work in part by targeting the genomically stable endothelial cell compartment, rather than the genomically unstable tumor cell compartment. New blood vessel formation is a relatively fragile process, subject to disruptive interference at several levels. In short, the therapy is the selection agent which is being used to kill a cell compartment. Tumor cells evolve resistance rapidly due to rapid generation time (days) and genomic instability (variation), whereas endothelial cells are a good target because of a long generation time (months) and genomic stability (low variation).
This is an example of selection in action at the cellular level, using a selection pressure to target and differentiate between varying populations of cells. The end result is the extinction of one species or population of cells (endothelial cells), followed by the collapse of the ecosystem (the tumor) due either to nutrient deprivation or self-pollution from the destruction of necessary waste pathways.
Angiogenesis-based tumor therapy relies on natural and synthetic angiogenesis inhibitors like angiostatin, endostatin and tumstatin. These are proteins that mainly originate as specific fragments of pre-existing structural proteins like collagen or plasminogen.
Recently, the first FDA-approved therapy targeted at angiogenesis in cancer came on the market in the US. This is a monoclonal antibody directed against an isoform of VEGF. The commercial name of this antibody is Avastin, and the therapy has been approved for use in colorectal cancer in combination with established chemotherapy.
Angiogenesis for cardiovascular disease
Angiogenesis represents an excellent therapeutic target for the treatment of cardiovascular disease. It is a potent, physiological process that underlies the natural manner in which our bodies respond to a diminution of blood supply to vital organs, namely the production of new collateral vessels to overcome the ischemic insult.[11] A large number of preclinical studies have been performed with protein-, gene- and cell-based therapies in animal models of cardiac ischemia, as well as models of peripheral artery disease. Reproducible and credible successes in these early animal studies led to high enthusiasm that this new therapeutic approach could be rapidly translated to a clinical benefit for millions of patients in the Western world suffering from these disorders. A decade of clinical testing both gene- and protein-based therapies designed to stimulate angiogenesis in underperfused tissues and organs, however, has led from one disappointment to another. Although all of these preclinical readouts, which offered great promise for the transition of angiogenesis therapy from animals to humans, were in one fashion or another, incorporated into early stage clinical trials, the FDA has, to date (2007), insisted that the primary endpoint for approval of an angiogenic agent must be an improvement in exercise performance of treated patients.
If one reviews in detail the various published angiogenesis clinical trials, it can be realized that most of these trials had success in achieving various secondary or supportive endpoints, but failed when attempting to demonstrate a statistically significant improvement in exercise performance, typically done by a treadmill exercise test.[37] Perhaps the greatest reason for these trials' failure to achieve success is the high occurrence of the "placebo effect" in studies employing treadmill exercise test readout. Thus, even though a majority of the treated patients in these trials experience relief of such clinical symptoms as chest pain (angina), and generally performed better on most efficacy readouts, there were enough "responders" in the blinded placebo groups to render the trial inconclusive. In addition to the placebo effect, more recent animal studies have also highlighted various factors that may inhibit an angiogenesis response, including certain drugs, smoking, and hypercholesterolemia.
Although shown to be relatively safe therapies, no angiogenic therapeutic has yet made it through the gauntlet of clinical testing required for drug approval. By capitalizing on the large database of what did and did not work in previous clinical trials, results from more recent studies with redesigned clinical protocols give renewed hope that angiogenesis therapy will be a treatment choice for sufferers of cardiovascular disease resulting from occluded and/or stenotic vessels.
Early clinical studies with protein-based therapeutics largely focused on the intravenous or intracoronary administration of a particular growth factor to stimulate angiogenesis in the affected tissue or organ. Most of these trials did not achieve statistically significant improvements in their clinical endpoints. This ultimately led to an abandonment of this approach and a widespread belief in the field that protein therapy, especially with a single agent, was not a viable option to treat ischemic cardiovascular disease. However, the failure of gene- or cell-based therapy to deliver a suitable treatment choice for diseases resulting from poor blood flow has led to a resurgence of interest in returning to protein-based therapy to stimulate angiogenesis.
These failures suggested that either these are the wrong molecular targets to induce neovascularization, that they can only be effectively used if formulated and administered correctly, or that their presentation in the context of the overall cellular microenvironment may play a vital role in their utility. It may be necessary to present these proteins in a way that mimics natural signaling events, including the concentration, spatial and temporal profiles, and their simultaneous or sequential presentation with other appropriate factors.[38]
Lessons learned from earlier protein-based studies, which indicated that intravenous or intracoronary delivery of the protein was not efficacious, have led to completed and ongoing clinical trials in which the angiogenic protein is injected directly into the beating ischemic heart.
Such localized administration of the potent angiogenic growth factor, human FGF-1, has recently given promising results in clinical trials in no-option heart patients.[15][16] Angiogenesis was documented by angiographically visible "blushing", and functional exercise tests were also performed on a subset of patients. The attractiveness of protein therapy is that large amounts of the therapeutic agent can be injected into the ischemic area of interest, to pharmacologically start the process of blood vessel growth and collateral arteries formation.[14] In addition, from pharmacokinetic data collected from the recent FGF-1 studies in the human heart, it appears that FGF-1, once it exits the heart, is cleared in less than three hours from the circulation. This would presumably prevent FGF-1 from stimulating unwanted angiogenesis in other tissues of the bodies where it could potentially cause harm, such as the retina and in the kidneys. No serious adverse events have yet to be noted in any of the completed or ongoing clinical trials in which the FGF-1 protein is used as the therapeutic agent to stimulate angiogenesis.[14][39]

Left: Angiographic "blushing" following FGF-1 injection into the human heart. Right, measurements of pixel density in angiograms ("gray-value-analysis") indicating a threefold increase in vessel density in the treated human myocardium (3 months and 3 years).
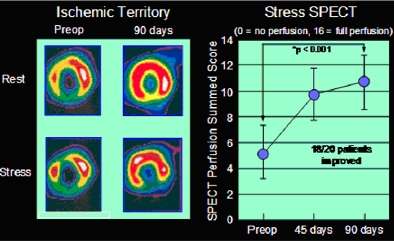
Improvement in myocardial perfusion (blood supply) after FGF-1 treatment as demonstrated by single photon emission computed tomography (SPECT) imaging.
Another approach for stimulation angiogenesis in the heart is using FGF gene (instead of the protein) delivered by adenoviral vectors (e.g. Ad5FGF-4, Generx) during intracoronary infusion. Several clinical trials with angiogenic gene therapy have been completed with promising efficacy and safety data (e.g. AGENT trials). These trials tested intracoronary administration of Ad5FGF-4 in over 450 patients and demonstrated the safety of the product and the procedure. It is notable that when exercise tolerance was used as the primary end-point, the product did not achieve statistically significant effect primarily due to high placebo effect in the control groups. In contrast, when the more objective end-point of SPECT (measuring the extent of reversible myocardial ischemia) was used in the AGENT-2 clinical study, intracoronary administration of Ad5FGF4 resulted in a statistically significant therapeutic effect. Currently ongoing pivotal (Phase 3) clinical studies use myocardial perfusion (as measured by SPECT) as the primary end point.
Exercise
Angiogenesis is generally associated with aerobic exercise and endurance exercise. While arteriogenesis produces network changes that allow for a large increase in the amount of total flow in a network, angiogenesis causes changes that allow for greater nutrient delivery over a long period of time. Capillaries are designed to provide maximum nutrient delivery efficiency, so an increase in the number of capillaries allows the network to deliver more nutrients in the same amount of time. A greater number of capillaries also allows for greater oxygen exchange in the network. This is vitally important to endurance training, because it allows a person to continue training for an extended period of time. However, no experimental evidence suggests that increased capillarity is required in endurance exercise to increase the maximum oxygen delivery.[7]
Macular degeneration
Overexpression of VEGF causes increased permeability in blood vessels in addition to stimulating angiogenesis. In wet macular degeneration, VEGF causes proliferation of capillaries into the retina. Since the increase in angiogenesis also causes edema, blood and other retinal fluids leak into the retina, causing loss of vision. Anti-angiogenic drugs targeting the VEGF pathways are now used successfully to treat this type of macular degeneration
Quantification
Quantifying vasculature parameters such as microvascular density has various complications due to preferential staining or limited representation of tissues by histological sections. Recent research has shown complete 3D reconstruction of tumor vascular structure and quantification of vessel structures in whole tumors in animal models.[40] This has been applied to assessing effects of chemotherapeutic drugs.
See also
References
- ↑ Birbrair, Alexander; Zhang, Tan; Wang, Zhong-Min; Messi, Maria Laura; Mintz, Akiva; Delbono, Osvaldo (2015-01-01). "Pericytes at the intersection between tissue regeneration and pathology". Clinical Science 128 (2): 81–93. doi:10.1042/CS20140278. ISSN 0143-5221. PMC 4200531. PMID 25236972.
- ↑ Birbrair, Alexander; Zhang, Tan; Wang, Zhong-Min; Messi, Maria Laura; Olson, John D.; Mintz, Akiva; Delbono, Osvaldo (2014-07-01). "Type-2 pericytes participate in normal and tumoral angiogenesis". American Journal of Physiology. Cell Physiology 307 (1): C25–C38. doi:10.1152/ajpcell.00084.2014. ISSN 0363-6143. PMC 4080181. PMID 24788248.
- ↑ Risau W, Flamme I (1995). "Vasculogenesis.". Annual review of cell and developmental biology 11: 73–91. doi:10.1146/annurev.cb.11.110195.000445. PMID 8689573.
- ↑ Flamme I, Frölich T, Risau W (November 1997). "Molecular mechanisms of vasculogenesis and embryonic angiogenesis". Journal of cellular physiology 173 (2): 206–10. doi:10.1002/(SICI)1097-4652(199711)173:2<206::AID-JCP22>3.0.CO;2-C. PMID 9365523.
- ↑ John S. Penn (11 March 2008). Retinal and Choroidal Angiogenesis. Springer. pp. 119–. ISBN 978-1-4020-6779-2. Retrieved 26 June 2010.
- ↑ Burri PH, Hlushchuk R, Djonov V (2004). "Intussusceptive angiogenesis: its emergence, its characteristics, and its significance". Dev Dyn. 231 (3): 474–88. doi:10.1002/dvdy.20184. PMID 15376313.
- 1 2 3 4 5 Prior BM, Yang HT, Terjung RL (September 2004). "What makes vessels grow with exercise training?". Journal of Applied Physiology 97 (3): 1119–28. doi:10.1152/japplphysiol.00035.2004. PMID 15333630.
- ↑ Perhaps an inhibitor of angiogenesis: Sheppard D (October 2002). "Endothelial integrins and angiogenesis: not so simple anymore". The Journal of Clinical Investigation 110 (7): 913–4. doi:10.1172/JCI16713. PMC 151161. PMID 12370267.
- ↑ Ornitz DM, Itoh N (2001). "Fibroblast growth factors". Genome Biology 2 (3): reviews3005.1–reviews3005.12. doi:10.1186/gb-2001-2-3-reviews3005. PMC 138918. PMID 11276432. Archived from the original on February 2, 2006.
- ↑ Blaber M, DiSalvo J, Thomas KA (February 1996). "X-ray crystal structure of human acidic fibroblast growth factor". Biochemistry 35 (7): 2086–94. doi:10.1021/bi9521755. PMID 8652550.
- 1 2 3 Stegmann TJ (December 1998). "FGF-1: a human growth factor in the induction of neoangiogenesis". Expert Opinion on Investigational Drugs 7 (12): 2011–5. doi:10.1517/13543784.7.12.2011. PMID 15991943.
- ↑ Khurana R, Simons M (April 2003). "Insights from angiogenesis trials using fibroblast growth factor for advanced arteriosclerotic disease". Trends in Cardiovascular Medicine 13 (3): 116–22. doi:10.1016/S1050-1738(02)00259-1. PMID 12691676.
- 1 2 Stegmann TJ, Hoppert T, Schneider A, Gemeinhardt S, Köcher M, Ibing R, Strupp G (September 2000). "Induction of myocardial neoangiogenesis by human growth factors. A new therapeutic approach in coronary heart disease". Herz (in German) 25 (6): 589–99. doi:10.1007/PL00001972. PMID 11076317.
- 1 2 3 4 Wagoner, L.E., Merrill, W., Jacobs, J., Conway, G., Boehmer, J., Thomas, K., Stegmann, T.J.: Angiogenesis Protein Therapy With Human Fibroblast Growth Factor (FGF-1) Results of a Phase I Open Label, Dose Escalation Study in Subjects With CAD Not Eligible For PCI Or CABG" Circulation 116: 443, 2007
- 1 2 Stegmann, T.J., Hoppert, T., Schneider, A., Popp, M., Strupp, G., Ibing, R.O., Hertel, A.: Therapeutic angiogenesis: intramyocardial growth factor delivery of FGF-1 as sole therapy in patients with chronic coronary artery disease. CVR. 2000; 1: 259-267
- 1 2 Wagoner, L.E., Snavely, D.D., Conway, G.A., Hauntz, E.A., Merrill, W.H.: Intramyocardial injection of fibroblast growth factor-1 for treatment of refractory angina pectoris: the initial US experience" Circulation 2004; 110: 395.
- ↑ Goto F, Goto K, Weindel K, Folkman J (November 1993). "Synergistic effects of vascular endothelial growth factor and basic fibroblast growth factor on the proliferation and cord formation of bovine capillary endothelial cells within collagen gels". Laboratory Investigation 69 (5): 508–17. PMID 8246443.
- ↑ Ding YH, Luan XD, Li J, Rafols JA, Guthinkonda M, Diaz FG, Ding Y (December 2004). "Exercise-induced overexpression of angiogenic factors and reduction of ischemia/reperfusion injury in stroke". Current Neurovascular Research 1 (5): 411–20. doi:10.2174/1567202043361875. PMID 16181089. Archived from the original on April 19, 2012.
- ↑ Gavin TP, Robinson CB, Yeager RC, England JA, Nifong LW, Hickner RC (January 2004). "Angiogenic growth factor response to acute systemic exercise in human skeletal muscle". Journal of Applied Physiology 96 (1): 19–24. doi:10.1152/japplphysiol.00748.2003. PMID 12949011.
- ↑ Kraus RM, Stallings HW, Yeager RC, Gavin TP (April 2004). "Circulating plasma VEGF response to exercise in sedentary and endurance-trained men". Journal of Applied Physiology 96 (4): 1445–50. doi:10.1152/japplphysiol.01031.2003. PMID 14660505.
- ↑ Lloyd PG, Prior BM, Yang HT, Terjung RL (May 2003). "Angiogenic growth factor expression in rat skeletal muscle in response to exercise training". American Journal of Physiology. Heart and Circulatory Physiology 284 (5): H1668–78. doi:10.1152/ajpheart.00743.2002 (inactive 2015-01-09). PMID 12543634.
- ↑ Thurston G (October 2003). "Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis". Cell and Tissue Research 314 (1): 61–8. doi:10.1007/s00441-003-0749-6. PMID 12915980.
- ↑ Haas TL, Milkiewicz M, Davis SJ, Zhou AL, Egginton S, Brown MD, Madri JA, Hudlicka O (October 1, 2000). "Matrix metalloproteinase activity is required for activity-induced angiogenesis in rat skeletal muscle". American Journal of Physiology. Heart and Circulatory Physiology 279 (4): H1540–7. PMID 11009439.
- ↑ Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ (February 2007). "Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting". Proceedings of the National Academy of Sciences of the United States of America 104 (9): 3219–24. Bibcode:2007PNAS..104.3219L. doi:10.1073/pnas.0611206104. PMC 1805530. PMID 17296940.
- ↑ Ferrara N, Kerbel RS (Dec 15, 2005). "Angiogenesis as a therapeutic target". Nature 438 (7070): 967–74. Bibcode:2005Natur.438..967F. doi:10.1038/nature04483. PMID 16355214.
- ↑ Folkman J, Klagsbrun M (January 1987). "Angiogenic factors". Science 235 (4787): 442–7. Bibcode:1987Sci...235..442F. doi:10.1126/science.2432664. PMID 2432664.
- ↑ Folkman J (September 1996). "Fighting cancer by attacking its blood supply". Scientific American 275 (3): 150–4. doi:10.1038/scientificamerican0996-150. PMID 8701285.
- ↑ Folkman J (February 24, 1998). "Angiogenic therapy of the human heart". Circulation 97 (7): 628–9. doi:10.1161/01.CIR.97.7.628. PMID 9495294.
- ↑ McDougall, S.R, Anderson, A.R.A., Chaplain, M.A.J. Mathematical modelling of dynamic adaptive tumour-induced angiogenesis: Clinical implications and therapeutic targeting strategies. Journal of Theoretical Biology 241. 2006. (Cites Folkman, 1971)
- ↑ Gonzalez-Perez, Ruben R.; Rueda, Bo R. (2013). Tumor angiogenesis regulators (1 ed.). Boca Raton: Taylor & Francis. p. 347. ISBN 978-1466580978. Retrieved 2 October 2014.
- ↑ Enzyme eliminated by cancer cells holds promise for cancer treatment
- ↑ Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, Tibbe AG, Uhr JW, Terstappen LW (October 2004). "Tumor Cells Circulate in the Peripheral Blood of All Major Carcinomas but not in Healthy Subjects or Patients With Nonmalignant Diseases". Clin. Cancer Research 10 (20): 6897–6904. doi:10.1158/1078-0432.CCR-04-0378. PMID 15501967.
- ↑ Bagri A, Kouros-Mehr H, Leong KG, Plowman GD (2010). "Use of anti-VEGF adjuvant therapy in cancer: challenges and rationale". Trends Mol Med. 16 (3): 122–32. doi:10.1016/j.molmed.2010.01.004. PMID 20189876.
- ↑ Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V (November 2010). "Glioblastoma stem-like cells give rise to tumour endothelium". Nature 468 (7325): 829–33. Bibcode:2010Natur.468..829W. doi:10.1038/nature09624. PMID 21102433.
- ↑ Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R (November 2010). "Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells". Nature 468 (7325): 824–8. Bibcode:2010Natur.468..824R. doi:10.1038/nature09557. PMID 21102434.
- ↑ Daring Tumors: Tumor Cells Make Their Own Blood Vessels http://www.sciguru.com/newsitem/5082/Daring-Tumors-Tumor-Cells-Make-Their-Own-Blood-Vessels/
- ↑ Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, Laham RJ, Li W, Pike M, Sellke FW, Stegmann TJ, Udelson JE, Rosengart TK (September 12, 2000). "Clinical trials in coronary angiogenesis: issues, problems, consensus: An expert panel summary". Circulation 102 (11): E73–86. doi:10.1161/01.CIR.102.11.e73. PMID 10982554.
- ↑ Cao L, Mooney DJ (November 2007). "Spatiotemporal control over growth factor signaling for therapeutic neovascularization". Advanced Drug Delivery Reviews 59 (13): 1340–50. doi:10.1016/j.addr.2007.08.012. PMC 2581871. PMID 17868951.
- ↑ Stegmann TJ (2005). New vessels for the heart : angiogenesis as new treatment for coronary heart (1st ed.). Henderson, NV: CardioVascular Biotherapies Inc. ISBN 978-0-9765583-0-9.
- ↑ Chia-Chi Chien, Ivan M. Kempson, Cheng Liang Wang et al, "Complete microscale profiling of tumor microangiogenesis" Biotechnology Advances (2011), doi:10.1016/j.biotechadv.2011.12.001.
External links
- Attacking Tumor Vasculature, A cure for Cancer. PMAP The Proteolysis Map-animation
- Angiogenesis for Heart Disease from Angioplasty.Org
- What You Need to Get Started Researching Your Options from CancerGuide
- Angiogenesis - The Virtual Library of Biochemistry and Cell Biology
- Visualizing Angiogenesis with GFP
- NCI Understanding Cancer series on Angiogenesis
- TherapeuticAngiogenesis.com
- Angiogenesis Foundation
- "Anti-fungal drug stops blood vessel growth". EurekAlert. 2007-04-27.
- Iris Pharma : animal models of Ocular Angiogenesis / Neovascularization
- Image analysis of angiogenesis with digital pathology
- William Li: Can we eat to starve cancer?
- Tumors Beware, Part 2 (from Beaker Blog)
- Angiogenesis - Thomas H. Adair and Jean-Pierre Montani. A textbook on the topic freely available at NCBI.
- Animation by Random42 Medical Animation Characteristics of angiogenic blood vessels in cancer
| ||||||||||
| ||||||||||||||||||||||||||||||||||||||