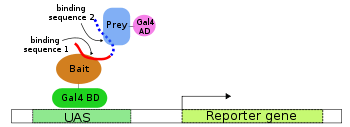
Two-hybrid screening

A. Gal4 transcription factor gene produces two domain protein (BD and AD), which is essential for transcription of the reporter gene (LacZ).
B,C. Two fusion proteins are prepared: Gal4BD+Bait and Gal4AD+Prey. None of them is usually sufficient to initiate the transcription (of the reporter gene) alone.
D. When both fusion proteins are produced and Bait part of the first interact with Prey part of the second, transcription of the reporter gene occurs.
Two-hybrid screening (also known as yeast two-hybrid system or Y2H) is a molecular biology technique used to discover protein–protein interactions (PPIs) [1] and protein–DNA interactions[2][3] by testing for physical interactions (such as binding) between two proteins or a single protein and a DNA molecule, respectively.
The premise behind the test is the activation of downstream reporter gene(s) by the binding of a transcription factor onto an upstream activating sequence (UAS). For two-hybrid screening, the transcription factor is split into two separate fragments, called the binding domain (BD) and activating domain (AD). The BD is the domain responsible for binding to the UAS and the AD is the domain responsible for the activation of transcription.[1][2] The Y2H is thus a protein-fragment complementation assay.
History
Pioneered by Stanley Fields and Ok-Kyu Song in 1989, the technique was originally designed to detect protein–protein interactions using the GAL4 transcriptional activator of the yeast Saccharomyces cerevisiae. The GAL4 protein activated transcription of a protein involved in galactose utilization, which formed the basis of selection.[4] Since then, the same principle has been adapted to describe many alternative methods, including some that detect protein–DNA interactions or DNA-DNA interactions, as well as methods that use Escherichia coli instead of yeast.[3]
Basic premise
The key to the two-hybrid screen is that in most eukaryotic transcription factors, the activating and binding domains are modular and can function in proximity to each other without direct binding.[5] This means that even though the transcription factor is split into two fragments, it can still activate transcription when the two fragments are indirectly connected.
The most common screening approach is the yeast two-hybrid assay.[6] This system often utilizes a genetically engineered strain of yeast in which the biosynthesis of certain nutrients (usually amino acids or nucleic acids) is lacking. When grown on media that lacks these nutrients, the yeast fail to survive. This mutant yeast strain can be made to incorporate foreign DNA in the form of plasmids. In yeast two-hybrid screening, separate bait and prey plasmids are simultaneously introduced into the mutant yeast strain.
Plasmids are engineered to produce a protein product in which the DNA-binding domain (BD) fragment is fused onto a protein while another plasmid is engineered to produce a protein product in which the activation domain (AD) fragment is fused onto another protein. The protein fused to the BD may be referred to as the bait protein, and is typically a known protein the investigator is using to identify new binding partners. The protein fused to the AD may be referred to as the prey protein and can be either a single known protein or a library of known or unknown proteins. In this context, a library may consist of a collection of protein-encoding sequences that represent all the proteins expressed in a particular organism or tissue, or may be generated by synthesising random DNA sequences.[3] Regardless of the source, they are subsequently incorporated into the protein-encoding sequence of a plasmid, which is then transfected into the cells chosen for the screening method.[3] This technique, when using a library, assumes that each cell is transfected with no more than a single plasmid and that, therefore, each cell ultimately expresses no more than a single member from the protein library.
If the bait and prey proteins interact (i.e., bind), then the AD and BD of the transcription factor are indirectly connected, bringing the AD in proximity to the transcription start site and transcription of reporter gene(s) can occur. If the two proteins do not interact, there is no transcription of the reporter gene. In this way, a successful interaction between the fused protein is linked to a change in the cell phenotype.[1]
The challenge of separating cells that express proteins that happen to interact with their counterpart fusion proteins from those that do not, is addressed in the following section.
Fixed domains
In any study, some of the protein domains, those under investigation, will be varied according to the goals of the study whereas other domains, those that are not themselves being investigated, will be kept constant. For example in a two-hybrid study to select DNA-binding domains, the DNA-binding domain, BD, will be varied while the two interacting proteins, the bait and prey, must be kept constant to maintain a strong binding between the BD and AD. There are a number of domains from which to choose the BD, bait and prey and AD, if these are to remain constant. In protein–protein interaction investigations, the BD may be chosen from any of many strong DNA-binding domains such as Zif268.[2] A frequent choice of bait and prey domains are residues 263–352 of yeast Gal11P with a N342V mutation[2] and residues 58–97 of yeast Gal4,[2] respectively. These domains can be used in both yeast- and bacterial-based selection techniques and are known to bind together strongly.[1][2]
The AD chosen must be able to activate transcription of the reporter gene, using the cell's own transcription machinery. Thus, the variety of ADs available for use in yeast-based techniques may not be suited to use in their bacterial-based analogues. The herpes simplex virus-derived AD, VP16 and yeast Gal4 AD have been used with success in yeast[1] whilst a portion of the α-subunit of E. coli RNA polymerase has been utilised in E. coli-based methods.[2][3]
Whilst powerfully activating domains may allow greater sensitivity towards weaker interactions, conversely, a weaker AD may provide greater stringency.
Construction of expression plasmids
A number of engineered genetic sequences must be incorporated into the host cell to perform two-hybrid analysis or one of its derivative techniques. The considerations and methods used in the construction and delivery of these sequences differ according to the needs of the assay and the organism chosen as the experimental background.
There are two broad categories of hybrid library: random libraries and cDNA-based libraries. A cDNA library is constituted by the cDNA produced through reverse transcription of mRNA collected from specific cells of types of cell. This library can be ligated into a construct so that it is attached to the BD or AD being used in the assay.[1] A random library uses lengths of DNA of random sequence in place of these cDNA sections. A number of methods exist for the production of these random sequences, including cassette mutagenesis.[2] Regardless of the source of the DNA library, it is ligated into the appropriate place in the relevant plasmid/phagemid using the appropriate restriction endonucleases.[2]
E. coli-specific considerations
By placing the hybrid proteins under the control of IPTG-inducible lac promoters, they are expressed only on media supplemented with IPTG. Further, by including different antibiotic resistance genes in each genetic construct, the growth of non-transformed cells is easily prevented through culture on media containing the corresponding antibiotics. This is particularly important for counter selection methods in which a lack of interaction is needed for cell survival.[2]
The reporter gene may be inserted into the E. coli genome by first inserting it into an episome, a type of plasmid with the ability to incorporate itself into the bacterial cell genome[2] with a copy number of approximately one per cell.[7]
The hybrid expression phagemids can be electroporated into E. coli XL-1 Blue cells which after amplification and infection with VCS-M13 helper phage, will yield a stock of library phage. These phage will each contain one single-stranded member of the phagemid library.[2]
Recovery of protein information
Once the selection has been performed, the primary structure of the proteins which display the appropriate characteristics must be determined. This is achieved by retrieval of the protein-encoding sequences (as originally inserted) from the cells showing the appropriate phenotype.
E. coli
The phagemid used to transform E. coli cells may be "rescued" from the selected cells by infecting them with VCS-M13 helper phage. The resulting phage particles that are produced contain the single-stranded phagemids and are used to infect XL-1 Blue cells.[2] The double-stranded phagemids are subsequently collected from these XL-1 Blue cells, essentially reversing the process used to produce the original library phage. Finally, the DNA sequences are determined through dideoxy sequencing.[2]
Controlling sensitivity
The Escherichia coli-derived Tet-R repressor can be used in line with a conventional reporter gene and can be controlled by tetracycline or doxicycline (Tet-R inhibitors). Thus the expression of Tet-R is controlled by the standard two-hybrid system but the Tet-R in turn controls (represses) the expression of a previously mentioned reporter such as HIS3, through its Tet-R promoter. Tetracycline or its derivatives can then be used to regulate the sensitivity of a system utilising Tet-R.[1]
Sensitivity may also be controlled by varying the dependency of the cells on their reporter genes. For example, this may be affected by altering the concentration of histidine in the growth medium for his3-dependent cells and altering the concentration of streptomycin for aadA dependent cells.[2][3] Selection-gene-dependency may also be controlled by applying an inhibitor of the selection gene at a suitable concentration. 3-Amino-1,2,4-triazole (3-AT) for example, is a competitive inhibitor of the HIS3-gene product and may be used to titrate the minimum level of HIS3 expression required for growth on histidine-deficient media.[2]
Sensitivity may also be modulated by varying the number of operator sequences in the reporter DNA.
Non-fusion proteins
A third, non-fusion protein may be co-expressed with two fusion proteins. Depending on the investigation, the third protein may modify one of the fusion proteins or mediate or interfere with their interaction.[1]
Co-expression of the third protein may be necessary for modification or activation of one or both of the fusion proteins. For example S. cerevisiae possesses no endogenous tyrosine kinase. If an investigation involves a protein that requires tyrosine phosphorylation, the kinase must be supplied in the form of a tyrosine kinase gene.[1]
The non-fusion protein may mediate the interaction by binding both fusion proteins simultaneously, as in the case of ligand-dependent receptor dimerization.[1]
For a protein with an interacting partner, its functional homology to other proteins may be assessed by supplying the third protein in non-fusion form, which then may or may not compete with the fusion-protein for its binding partner. Binding between the third protein and the other fusion protein will interrupt the formation of the reporter expression activation complex and thus reduce reporter expression, leading to the distinguishing change in phenotype.[1]
Split-ubiquitin yeast two-hybrid
One limitation of classic yeast two-hybrid screens is that they are limited to soluble proteins. It is therefore impossible to use them to study the protein–protein interactions between insoluble integral membrane proteins. The split-ubiquitin system provides a method for overcoming this limitation.[8] In the split-ubiquitin system, two integral membrane proteins to be studied are fused to two different ubiquitin moieties: a C-terminal ubiquitin moiety ("Cub", residues 35–76) and an N-terminal ubiquitin moiety ("Nub", residues 1–34). These fused proteins are called the bait and prey, respectively. In addition to being fused to an integral membrane protein, the Cub moiety is also fused to a transcription factor (TF) that can be cleaved off by ubiquitin specific proteases. Upon bait–prey interaction, Nub and Cub-moieties assemble, reconstituting the split-ubiquitin. The reconstituted split-ubiquitin molecule is recognized by ubiquitin specific proteases, which cleave off the transcription factor, allowing it to induce the transcription of reporter genes.[9]
Fluorescent Two-hybrid Assay
Zolghadr and co-workers presented a fluorescent two-hybrid system that uses two hybrid proteins that are fused to different fluorescent proteins as well as LacI, the lac repressor. The structure of the fusion proteins looks like this: FP2-LacI-bait and FP1-prey where the bait and prey proteins interact and bring the fluorescent proteins (FP1 = GFP, FP2=mCherry) in close proximity at the binding site of the LacI protein in the host cell genome.[10] The system can also be used to screen for inhibitors of protein-protein interactions.[11]
Enzymatic two-hybrid systems: KISS
While the original Y2H system used a reconstituted transcription factor, other systems create enzymatic activities to detect PPIs. For instance, the KInase Substrate Sensor ("KISS"), is a mammalian two-hybrid approach has been designed to map intracellular PPIs. Here, a bait protein is fused to a kinase-containing portion of TYK2 and a prey is coupled to a gp130 cytokine receptor fragment. When bait and prey interact, TYK2 phosphorylates STAT3 docking sites on the prey chimera, which ultimately leads to activation of a reporter gene.[12]
One-, three- and one-two-hybrid variants
One-hybrid
The one-hybrid variation of this technique is designed to investigate protein–DNA interactions and uses a single fusion protein in which the AD is linked directly to the binding domain. The binding domain in this case however is not necessarily of fixed sequence as in two-hybrid protein–protein analysis but may be constituted by a library. This library can be selected against the desired target sequence, which is inserted in the promoter region of the reporter gene construct. In a positive-selection system, a binding domain that successfully binds the UAS and allows transcription is thus selected.[1]
Note that selection of DNA-binding domains is not necessarily performed using a one-hybrid system, but may also be performed using a two-hybrid system in which the binding domain is varied and the bait and prey proteins are kept constant.[2][3]
Three-hybrid
RNA-protein interactions have been investigated through a three-hybrid variation of the two-hybrid technique. In this case, a hybrid RNA molecule serves to adjoin together the two protein fusion domains—which are not intended to interact with each other but rather the intermediary RNA molecule (through their RNA-binding domains).[1] Techniques involving non-fusion proteins that perform a similar function, as described in the 'non-fusion proteins' section above, may also be referred to as three-hybrid methods.
One-two-hybrid
Simultaneous use of the one- and two-hybrid methods (that is, simultaneous protein–protein and protein–DNA interaction) is known as a one-two-hybrid approach and expected to increase the stringency of the screen.[1]
Host organism
Although theoretically, any living cell might be used as the background to a two-hybrid analysis, there are practical considerations that dictate which is chosen. The chosen cell line should be relatively cheap and easy to culture and sufficiently robust to withstand application of the investigative methods and reagents.[1]
Yeast
S. cerevisiae was the model organism used during the two-hybrid technique's inception. It has several characteristics that make it a robust organism to host the interaction, including the ability to form tertiary protein structures, neutral internal pH, enhanced ability to form disulfide bonds and reduced-state glutathione among other cytosolic buffer factors, to maintain a hospitable internal environment.[1] The yeast model can be manipulated through non-molecular techniques and its complete genome sequence is known.[1] Yeast systems are tolerant of diverse culture conditions and harsh chemicals that could not be applied to mammalian tissue cultures.[1]
A number of yeast strains have been created specifically for Y2H screens, e.g. Y187[13] and AH109,[14] both produced by Clontech. Yeast strains R2HMet and BK100 have also been used.[15]
E. coli
E. coli-based methods have several characteristics that may make them preferable to yeast-based homologues. The higher transformation efficiency and faster rate of growth lends E. coli to the use of larger libraries (in excess of 108).[2] A low false positive rate of approximately 3x10−8, the absence of requirement for a nuclear localisation signal to be included in the protein sequence and the ability to study proteins that would be toxic to yeast may also be major factors to consider when choosing an experimental background organism.[2]
It may be of note that the methylation activity of certain E. coli DNA methyltransferase proteins may interfere with some DNA-binding protein selections. If this is anticipated, the use of an E. coli strain that is defective for a particular methyltransferase may be an obvious solution.[2]
Applications
Determination of sequences crucial for interaction
By changing specific amino acids by mutating the corresponding DNA base-pairs in the plasmids used, the importance of those amino acid residues in maintaining the interaction can be determined.[1]
After using bacterial cell-based method to select DNA-binding proteins, it is necessary to check the specificity of these domains as there is a limit to the extent to which the bacterial cell genome can act as a sink for domains with an affinity for other sequences (or indeed, a general affinity for DNA).[2]
Drug and poison discovery
Protein–protein signalling interactions pose suitable therapeutic targets due to their specificity and pervasiveness. The random drug discovery approach uses compound banks that comprise random chemical structures, and requires a high-throughput method to test these structures in their intended target.[1]
The cell chosen for the investigation can be specifically engineered to mirror the molecular aspect that the investigator intends to study and then used to identify new human or animal therapeutics or anti-pest agents.[1]
Determination of protein function
By determination of the interaction partners of unknown proteins, the possible functions of these new proteins may be inferred.[1] This can be done using a single known protein against a library of unknown proteins or conversely, by selecting from a library of known proteins using a single protein of unknown function.[1]
Zinc finger protein selection
To select zinc finger proteins (ZFPs) for protein engineering, methods adapted from the two-hybrid screening technique have been used with success.[2][3] A ZFP is itself a DNA-binding protein used in the construction of custom DNA-binding domains that bind to a desired DNA sequence.[16]
By using a selection gene with the desired target sequence included in the UAS, and randomising the relevant amino acid sequences to produce a ZFP library, cells that host a DNA-ZFP interaction with the required characteristics can be selected. Each ZFP typically recognises only 3–4 base pairs, so to prevent recognition of sites outside the UAS, the randomised ZFP is engineered into a 'scaffold' consisting of another two ZFPs of constant sequence. The UAS is thus designed to include the target sequence of the constant scaffold in addition to the sequence for which a ZFP is selected.[2][3]
A number of other DNA-binding domains may also be investigated using this system.[2]
Strengths
- Two-hybrid screens are low-tech; they can be carried out in any lab without sophisticated equipment.
- Two-hybrid screens can provide an important first hint for the identification of interaction partners.
- The assay is scalable, which makes it possible to screen for interactions among many proteins. Furthermore, it can be automated, and by using robots many proteins can be screened against thousands of potentially interacting proteins in a relatively short time.
- Yeast two-hybrid data can be of similar quality to data generated by the alternative approach of coaffinity purification followed by mass spectrometry (AP/MS).[17]
Weaknesses
- The main criticism applied to the yeast two-hybrid screen of protein–protein interactions is the possibility of a high number of false positive (and false negative) identifications. The exact rate of false positive results is not known, but earlier estimates were as high as 70%.[18] The reason for this high error rate lies in the characteristics of the screen:
- Certain assay variants overexpress the fusion proteins which may cause unnatural protein concentrations that lead to unspecific (false) positives.
- The hybrid proteins are fusion proteins; that is, the fused parts may inhibit certain interactions, especially if an interaction takes place at the N-terminus of a test protein (where the DNA-binding or activation domain is typically attached).
- An interaction may not happen in yeast, the typical host organism for Y2H. For instance, if a bacterial protein is tested in yeast, it may lack a chaperone for proper folding that is only present in its bacterial host. Moreover, a mammalian protein is sometimes not correctly modified in yeast (e.g., missing phosphorylation), which can also lead to false results.
- The Y2H takes place in the nucleus. If test proteins are not localized to the nucleus (because they have other localization signals) two interacting proteins may be found to be non-interacting.
- Some proteins might specifically interact when they are co-expressed in the yeast, although in reality they are never present in the same cell at the same time. However, in most cases it cannot be ruled out that such proteins are indeed expressed in certain cells or under certain circumstances.
Each of these points alone can give rise to false results. Due to the combined effects of all error sources yeast two-hybrid have to be interpreted with caution. The probability of generating false positives means that all interactions should be confirmed by a high confidence assay, for example co-immunoprecipitation of the endogenous proteins, which is difficult for large scale protein–protein interaction data. Alternatively, Y2H data can be verified using multiple Y2H variants[19] or bioinformatics techniques. The latter test whether interacting proteins are expressed at the same time, share some common features (such as gene ontology annotations or certain network topologies), have homologous interactions in other species.[20]
See also
- Phage display, an alternative method for detecting protein–protein and protein–DNA interactions
- Protein array, a chip-based method for detecting protein–protein interactions
- Synthetic genetic array analysis, a yeast based method for studying gene interactions
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 Young K (1998). "Yeast two-hybrid: so many interactions, (in) so little time.". Biol Reprod 58 (2): 302–11. doi:10.1095/biolreprod58.2.302. PMID 9475380.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 Joung J, Ramm E, Pabo C (2000). "A bacterial two-hybrid selection system for studying protein-DNA and protein-protein interactions". Proc. Natl. Acad. Sci. U.S.A. 97 (13): 7382–7. Bibcode:2000PNAS...97.7382J. doi:10.1073/pnas.110149297. PMC 16554. PMID 10852947.
- 1 2 3 4 5 6 7 8 9 Hurt J, Thibodeau S, Hirsh A, Pabo C, Joung J (2003). "Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection". Proc. Natl. Acad. Sci. U.S.A. 100 (21): 12271–6. Bibcode:2003PNAS..10012271H. doi:10.1073/pnas.2135381100. PMC 218748. PMID 14527993.
- ↑ Fields S, Song O (1989). "A novel genetic system to detect protein-protein interactions" (abstract). Nature 340 (6230): 245–6. Bibcode:1989Natur.340..245F. doi:10.1038/340245a0. PMID 2547163. Abstract is free; full-text article is not.
- ↑ Verschure P, Visser A, Rots M (2006). "Step out of the groove: epigenetic gene control systems and engineered transcription factors". Adv Genet 56: 163–204. doi:10.1016/S0065-2660(06)56005-5. PMID 16735158.
- ↑ Gietz R.D., Triggs-Raine Barbara, Robbins Anne, Graham Kevin, Woods Robin (1997). "Identification of proteins that interact with a protein of interest: Applications of the yeast two-hybrid system". Mol Cel Biochem 172 (1–2): 67–79. doi:10.1023/A:1006859319926. PMID 9278233.
- ↑ Whipple F (1998). "Genetic analysis of prokaryotic and eukaryotic DNA-binding proteins in Escherichia coli". Nucleic Acids Res 26 (16): 3700–6. doi:10.1093/nar/26.16.3700. PMC 147751. PMID 9685485.
- ↑ Stagljar I, Korostensky C, Johnsson N, te Heesen S (April 1998). "A genetic system based on split-ubiquitin for the analysis of interactions between membrane proteins in vivo". Proc. Natl. Acad. Sci. U.S.A. 95 (9): 5187–92. Bibcode:1998PNAS...95.5187S. doi:10.1073/pnas.95.9.5187. PMC 20236. PMID 9560251.
- ↑ Snider, J; Kittanakom, S; Curak, J; Stagljar, I (2010). "Split-ubiquitin based membrane yeast two-hybrid (MYTH) system: A powerful tool for identifying protein-protein interactions". Journal of Visualized Experiments (36). doi:10.3791/1698. PMC 2818708. PMID 20125081.
- ↑ Zolghadr, K; Mortusewicz, O; Rothbauer, U; Kleinhans, R; Goehler, H; Wanker, E. E.; Cardoso, M. C.; Leonhardt, H (2008). "A fluorescent two-hybrid assay for direct visualization of protein interactions in living cells". Molecular & Cellular Proteomics 7 (11): 2279–87. doi:10.1074/mcp.M700548-MCP200. PMID 18622019.
- ↑ Yurlova, L; Derks, M; Buchfellner, A; Hickson, I; Janssen, M; Morrison, D; Stansfield, I; Brown, C. J.; Ghadessy, F. J.; Lane, D. P.; Rothbauer, U; Zolghadr, K; Krausz, E (2014). "The Fluorescent Two-Hybrid Assay to Screen for Protein-Protein Interaction Inhibitors in Live Cells: Targeting the Interaction of p53 with Mdm2 and Mdm4". Journal of Biomolecular Screening 19 (4): 516–25. doi:10.1177/1087057113518067. PMID 24476585.
- ↑ Lievens, S; Gerlo, S; Lemmens, I; De Clercq, D. J.; Risseeuw, M. D.; Vanderroost, N; De Smet, A. S.; Ruyssinck, E; Chevet, E; Van Calenbergh, S; Tavernier, J (2014). "KISS, a mammalian in situ protein interaction sensor". Molecular & Cellular Proteomics 13: 3332–42. doi:10.1074/mcp.M114.041087. PMID 25154561.
- ↑ Fromont-Racine, Micheline; Rain, Jean-Christophe; Legrain, Pierre (1997). "Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens". Nature Genetics 16 (3): 277–282. doi:10.1038/ng0797-277. PMID 9207794.
- ↑ Lu, L; Horstmann, H; Ng, C; Hong, W (December 2001). "Regulation of Golgi structure and function by ARF-like protein 1 (Arl1).". Journal of Cell Science 114 (Pt 24): 4543–55. PMID 11792819.
- ↑ Khadka, S.; Vangeloff, A. D.; Zhang, C.; Siddavatam, P.; Heaton, N. S.; Wang, L.; Sengupta, R.; Sahasrabudhe, S.; Randall, G.; Gribskov, M.; Kuhn, R. J.; Perera, R.; LaCount, D. J. (12 September 2011). "A Physical Interaction Network of Dengue Virus and Human Proteins". Molecular & Cellular Proteomics 10 (12): M111.012187–M111.012187. doi:10.1074/mcp.M111.012187.
- ↑ Gommans W, Haisma H, Rots M (2005). "Engineering zinc finger protein transcription factors: therapeutic relevance of switching endogenous gene expression on or off at command" (PDF). J Mol Biol 354 (3): 507–19. doi:10.1016/j.jmb.2005.06.082. PMID 16253273.
- ↑ Haiyuan Yu; et al. (2008). "High-Quality Binary Protein Interaction Map of the Yeast Interactome Network". Science 322 (5898): 104–110. Bibcode:2008Sci...322..104Y. doi:10.1126/science.1158684. PMC 2746753. PMID 18719252.
- ↑ Deane C, Salwiński Ł, Xenarios I, Eisenberg D (2002). "Protein interactions: two methods for assessment of the reliability of high throughput observations". Mol Cell Proteomics 1 (5): 349–56. doi:10.1074/mcp.M100037-MCP200. PMID 12118076.
- ↑ Chen, Y. C.; Rajagopala, S. V.; Stellberger, T.; Uetz, P. (2010). "Exhaustive benchmarking of the yeast two-hybrid system". Nature Methods 7 (9): 667–668; author 668 668. doi:10.1038/nmeth0910-667. PMID 20805792.
- ↑ Koegl, M.; Uetz, P. (2008). "Improving yeast two-hybrid screening systems". Briefings in Functional Genomics and Proteomics 6 (4): 302–312. doi:10.1093/bfgp/elm035. PMID 18218650.
External links
| Library resources about Two-hybrid screening |
- Complete Yeast Two-Hybrid Protocol
- Detail on sister technique two-hybrid system
- Science Creative Quarterly's overview of the yeast two hybrid system
- Gateway-Compatible Yeast One-Hybrid Screens
- Video animation of the Yeast Two-Hybrid System
- Two-Hybrid System Techniques at the US National Library of Medicine Medical Subject Headings (MeSH)