Tight binding
In solid-state physics, the tight-binding model (or TB model) is an approach to the calculation of electronic band structure using an approximate set of wave functions based upon superposition of wave functions for isolated atoms located at each atomic site. The method is closely related to the LCAO method (linear combination of atomic orbitals method) used in chemistry. Tight-binding models are applied to a wide variety of solids. The model gives good qualitative results in many cases and can be combined with other models that give better results where the tight-binding model fails. Though the tight-binding model is a one-electron model, the model also provides a basis for more advanced calculations like the calculation of surface states and application to various kinds of many-body problem and quasiparticle calculations.
Introduction
The name "tight binding" of this electronic band structure model suggests that this quantum mechanical model describes the properties of tightly bound electrons in solids. The electrons in this model should be tightly bound to the atom to which they belong and they should have limited interaction with states and potentials on surrounding atoms of the solid. As a result the wave function of the electron will be rather similar to the atomic orbital of the free atom to which it belongs. The energy of the electron will also be rather close to the ionization energy of the electron in the free atom or ion because the interaction with potentials and states on neighboring atoms is limited.
Though the mathematical formulation[1] of the one-particle tight-binding Hamiltonian may look complicated at first glance, the model is not complicated at all and can be understood intuitively quite easily. There are only three kinds of matrix elements that play a significant role in the theory. Two of those three kinds of elements should be close to zero and can often be neglected. The most important elements in the model are the interatomic matrix elements, which would simply be called the bond energies by a chemist.
In general there are a number of atomic energy levels and atomic orbitals involved in the model. This can lead to complicated band structures because the orbitals belong to different point-group representations. The reciprocal lattice and the Brillouin zone often belong to a different space group than the crystal of the solid. High-symmetry points in the Brillouin zone belong to different point-group representations. When simple systems like the lattices of elements or simple compounds are studied it is often not very difficult to calculate eigenstates in high-symmetry points analytically. So the tight-binding model can provide nice examples for those who want to learn more about group theory.
The tight-binding model has a long history and has been applied in many ways and with many different purposes and different outcomes. The model doesn't stand on its own. Parts of the model can be filled in or extended by other kinds of calculations and models like the nearly-free electron model. The model itself, or parts of it, can serve as the basis for other calculations.[2] In the study of conductive polymers, organic semiconductors and molecular electronics, for example, tight-binding-like models are applied in which the role of the atoms in the original concept is replaced by the molecular orbitals of conjugated systems and where the interatomic matrix elements are replaced by inter- or intramolecular hopping and tunneling parameters. These conductors nearly all have very anisotropic properties and sometimes are almost perfectly one-dimensional.
Historical background
By 1928, the idea of a molecular orbital had been advanced by Robert Mulliken, who was influenced considerably by the work of Friedrich Hund. The LCAO method for approximating molecular orbitals was introduced in 1928 by B. N. Finklestein and G. E. Horowitz, while the LCAO method for solids was developed by Felix Bloch, as part of his doctoral dissertation in 1928, concurrently with and independent of the LCAO-MO approach. A much simpler interpolation scheme for approximating the electronic band structure, especially for the d-bands of transition metals, is the parameterized tight-binding method conceived in 1954 by John Clarke Slater and George Fred Koster,[1] sometimes referred to as the SK tight-binding method. With the SK tight-binding method, electronic band structure calculations on a solid need not be carried out with full rigor as in the original Bloch's theorem but, rather, first-principles calculations are carried out only at high-symmetry points and the band structure is interpolated over the remainder of the Brillouin zone between these points.
In this approach, interactions between different atomic sites are considered as perturbations. There exist several kinds of interactions we must consider. The crystal Hamiltonian is only approximately a sum of atomic Hamiltonians located at different sites and atomic wave functions overlap adjacent atomic sites in the crystal, and so are not accurate representations of the exact wave function. There are further explanations in the next section with some mathematical expressions.
Recently, in the research about strongly correlated material, the tight binding approach is basic approximation because highly localized electrons like 3-d transition metal electrons sometimes display strongly correlated behaviors. In this case, the role of electron-electron interaction must be considered using the many-body physics description.
The tight-binding model is typically used for calculations of electronic band structure and band gaps in the static regime. However, in combination with other methods such as the random phase approximation (RPA) model, the dynamic response of systems may also be studied.
Mathematical formulation
We introduce the atomic orbitals 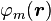 , which are eigenfunctions of the Hamiltonian
, which are eigenfunctions of the Hamiltonian 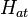 of a single isolated atom. When the atom is placed in a crystal, this atomic wave function overlaps adjacent atomic sites, and so are not true eigenfunctions of the crystal Hamiltonian. The overlap is less when electrons are tightly bound, which is the source of the descriptor "tight-binding". Any corrections to the atomic potential
of a single isolated atom. When the atom is placed in a crystal, this atomic wave function overlaps adjacent atomic sites, and so are not true eigenfunctions of the crystal Hamiltonian. The overlap is less when electrons are tightly bound, which is the source of the descriptor "tight-binding". Any corrections to the atomic potential 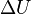 required to obtain the true Hamiltonian
required to obtain the true Hamiltonian 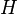 of the system, are assumed small:
of the system, are assumed small:
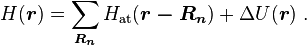
A solution 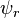 to the time-independent single electron Schrödinger equation is then approximated as a linear combination of atomic orbitals
to the time-independent single electron Schrödinger equation is then approximated as a linear combination of atomic orbitals 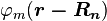 :
:
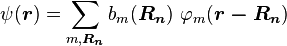 ,
,
where  refers to the m-th atomic energy level and
refers to the m-th atomic energy level and  locates an atomic site in the crystal lattice.
locates an atomic site in the crystal lattice.
Translational symmetry and normalization
The Bloch theorem states that the wave function in crystal can change under translation only by a phase factor:
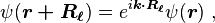
where  is the wave vector of the wave function. Consequently, the coefficients satisfy
is the wave vector of the wave function. Consequently, the coefficients satisfy
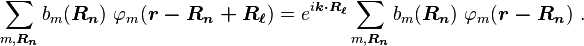
By substituting 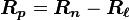 , we find
, we find
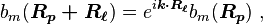 (where in RHS we have replaced the dummy index
(where in RHS we have replaced the dummy index 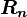 with
with 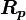 )
)
or
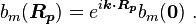
Normalizing the wave function to unity:
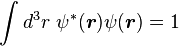
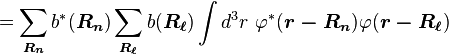
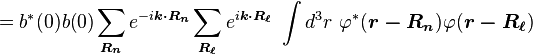
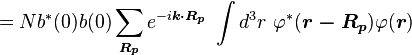
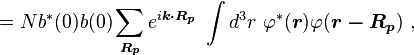
so the normalization sets b(0) as
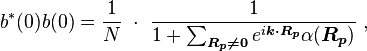
where α (Rp ) are the atomic overlap integrals, which frequently are neglected resulting in[3]
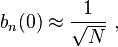
and
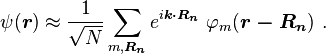
The tight binding Hamiltonian
Using the tight binding form for the wave function, and assuming only the m-th atomic energy level is important for the m-th energy band, the Bloch energies  are of the form
are of the form
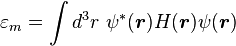
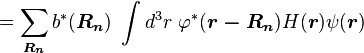
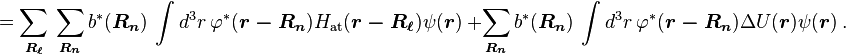
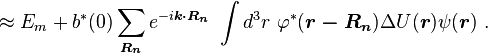
Here terms involving the atomic Hamiltonian at sites other than where it is centered are neglected. The energy then becomes
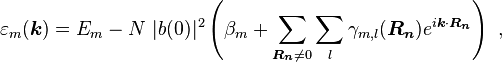
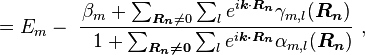
where Em is the energy of the m-th atomic level, and 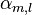 ,
, 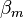 and
and 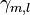 are the tight binding matrix elements.
are the tight binding matrix elements.
The tight binding matrix elements
The element
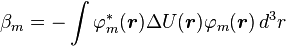 ,
,
is the atomic energy shift due to the potential on neighboring atoms. This term is relatively small in most cases. If it is large it means that potentials on neighboring atoms have a large influence on the energy of the central atom.
The next term
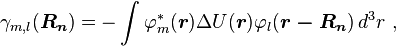
is the inter atomic matrix element between the atomic orbitals m and l on adjacent atoms. It is also called the bond energy or two center integral and it is the most important element in the tight binding model.
The last terms
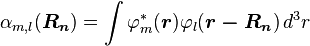 ,
,
denote the overlap integrals between the atomic orbitals m and l on adjacent atoms.
Evaluation of the matrix elements
As mentioned before the values of the -matrix elements are not so large in comparison with the ionization energy because the potentials of neighboring atoms on the central atom are limited. If is not relatively small it means that the potential of the neighboring atom on the central atom is not small either. In that case it is an indication that the tight binding model is not a very good model for the description of the band structure for some reason. The inter atomic distances can be too small or the charges on the atoms or ions in the lattice is wrong for example.
The inter atomic matrix elements can be calculated directly if the atomic wave functions and the potentials are known in detail. Most often this is not the case. There are numerous ways to get parameters for these matrix elements. Parameters can be obtained from chemical bond energy data. Energies and eigenstates on some high symmetry points in the Brillouin zone can be evaluated and values integrals in the matrix elements can be matched with band structure data from other sources.
The inter atomic overlap matrix elements should be rather small or neglectable. If they are large it is again an indication that the tight binding model is of limited value for some purposes. Large overlap is an indication for too short inter atomic distance for example. In metals and transition metals the broad s-band or sp-band can be fitted better to an existing band structure calculation by the introduction of next-nearest-neighbor matrix elements and overlap integrals but fits like that don't yield a very useful model for the electronic wave function of a metal. Broad bands in dense materials are better described by a nearly free electron model.
The tight binding model works particularly well in cases where the band width is small and the electrons are strongly localized, like in the case of d-bands and f-bands. The model also gives good results in the case of open crystal structures, like diamond or silicon, where the number of neighbors is small. The model can easily be combined with a nearly free electron model in a hybrid NFE-TB model.[2]
Connection to Wannier functions
Bloch wave functions describe the electronic states in a periodic crystal lattice. Bloch functions can be represented as a Fourier series[4]
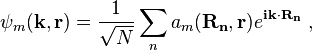
where Rn denotes an atomic site in a periodic crystal lattice, k is the wave vector of the Bloch wave, r is the electron position, m is the band index, and the sum is over all N atomic sites. The Bloch wave is an exact eigensolution for the wave function of an electron in a periodic crystal potential corresponding to an energy Em (k), and is spread over the entire crystal volume.
Using the Fourier transform analysis, a spatially localized wave function for the m-th energy band can be constructed from multiple Bloch waves:
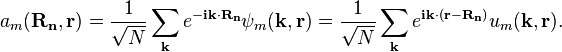
These real space wave functions 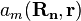 are called Wannier functions, and are fairly closely localized to the atomic site Rn. Of course, if we have exact Wannier functions, the exact Bloch functions can be derived using the inverse Fourier transform.
are called Wannier functions, and are fairly closely localized to the atomic site Rn. Of course, if we have exact Wannier functions, the exact Bloch functions can be derived using the inverse Fourier transform.
However it is not easy to calculate directly either Bloch functions or Wannier functions. An approximate approach is necessary in the calculation of electronic structures of solids. If we consider the extreme case of isolated atoms, the Wannier function would become an isolated atomic orbital. That limit suggests the choice of an atomic wave function as an approximate form for the Wannier function, the so-called tight binding approximation.
Second quantization
Modern explanations of electronic structure like t-J model and Hubbard model are based on tight binding model.[5] Tight binding can be understood by working under a second quantization formalism.
Using the atomic orbital as a basis state, the second quantization Hamiltonian operator in the tight binding framework can be written as:
-
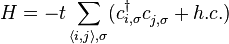 ,
, -
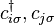 - creation and annihilation operators
- creation and annihilation operators
-
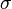 - spin polarization
- spin polarization
-
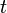 - hopping integral
- hopping integral
-
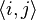 -nearest neighbor index
-nearest neighbor index
Here, hopping integral corresponds to the transfer integral 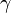 in tight binding model. Considering extreme cases of
in tight binding model. Considering extreme cases of 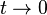 , it is impossible for an electron to hop into neighboring sites. This case is the isolated atomic system. If the hopping term is turned on (
, it is impossible for an electron to hop into neighboring sites. This case is the isolated atomic system. If the hopping term is turned on (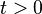 ) electrons can stay in both sites lowering their kinetic energy.
) electrons can stay in both sites lowering their kinetic energy.
In the strongly correlated electron system, it is necessary to consider the electron-electron interaction. This term can be written in
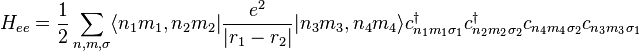
This interaction Hamiltonian includes direct Coulomb interaction energy and exchange interaction energy between electrons. There are several novel physics induced from this electron-electron interaction energy, such as metal-insulator transitions (MIT), high-temperature superconductivity, and several quantum phase transitions.
Example: one-dimensional s-band
Here the tight binding model is illustrated with a s-band model for a string of atoms with a single s-orbital in a straight line with spacing a and σ bonds between atomic sites.
To find approximate eigenstates of the Hamiltonian, we can use a linear combination of the atomic orbitals
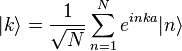
where N = total number of sites and  is a real parameter with
is a real parameter with 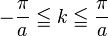 . (This wave function is normalized to unity by the leading factor 1/√N provided overlap of atomic wave functions is ignored.) Assuming only nearest neighbor overlap, the only non-zero matrix elements of the Hamiltonian can be expressed as
. (This wave function is normalized to unity by the leading factor 1/√N provided overlap of atomic wave functions is ignored.) Assuming only nearest neighbor overlap, the only non-zero matrix elements of the Hamiltonian can be expressed as
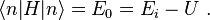
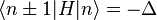
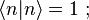
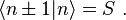
The energy Ei is the ionization energy corresponding to the chosen atomic orbital and U is the energy shift of the orbital as a result of the potential of neighboring atoms. The 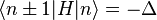 elements, which are the Slater and Koster interatomic matrix elements, are the bond energies
elements, which are the Slater and Koster interatomic matrix elements, are the bond energies 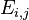 . In this one dimensional s-band model we only have
. In this one dimensional s-band model we only have 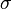 -bonds between the s-orbitals with bond energy
-bonds between the s-orbitals with bond energy 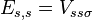 . The overlap between states on neighboring atoms is S. We can derive the energy of the state
. The overlap between states on neighboring atoms is S. We can derive the energy of the state 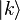 using the above equation:
using the above equation:
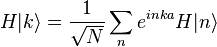
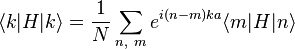
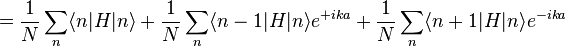
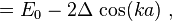
where, for example,
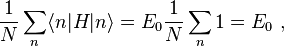
and
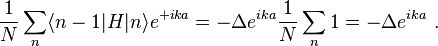
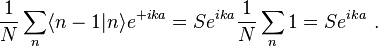
Thus the energy of this state can be represented in the familiar form of the energy dispersion:
 .
.
- For
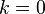 the energy is
the energy is 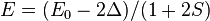 and the state consists of a sum of all atomic orbitals. This state can be viewed as a chain of bonding orbitals.
and the state consists of a sum of all atomic orbitals. This state can be viewed as a chain of bonding orbitals. - For
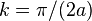 the energy is
the energy is 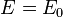 and the state consists of a sum of atomic orbitals which are a factor
and the state consists of a sum of atomic orbitals which are a factor 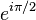 out of phase. This state can be viewed as a chain of non-bonding orbitals.
out of phase. This state can be viewed as a chain of non-bonding orbitals. - Finally for
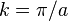 the energy is
the energy is 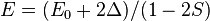 and the state consists of an alternating sum of atomic orbitals. This state can be viewed as a chain of anti-bonding orbitals.
and the state consists of an alternating sum of atomic orbitals. This state can be viewed as a chain of anti-bonding orbitals.
This example is readily extended to three dimensions, for example, to a body-centered cubic or face-centered cubic lattice by introducing the nearest neighbor vector locations in place of simply n a.[6] Likewise, the method can be extended to multiple bands using multiple different atomic orbitals at each site. The general formulation above shows how these extensions can be accomplished.
Table of interatomic matrix elements
In 1954 J.C. Slater and G.F. Koster published, mainly for the calculation of transition metal d-bands, a table of interatomic matrix elements[1]
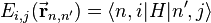
which, with a little patience and effort, can also be derived from the cubic harmonic orbitals straightforwardly. The table expresses the matrix elements as functions of LCAO two-centre bond integrals between two cubic harmonic orbitals, i and j, on adjacent atoms. The bond integrals are for example the 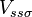 ,
, 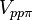 and
and 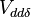 for sigma, pi and delta bonds.
for sigma, pi and delta bonds.
The interatomic vector is expressed as
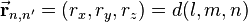
where d is the distance between the atoms and l, m and n are the direction cosines to the neighboring atom.
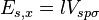
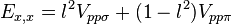
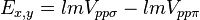
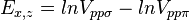
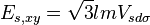
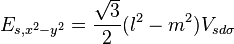
![E_{s,3z^2-r^2} = [n^2 - (l^2 + m^2) / 2] V_{sd\sigma}](../I/m/5669479a8dfa6d5d58d8ca3f41e67450.png)
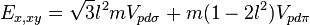
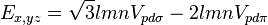
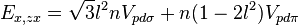
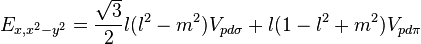
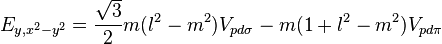
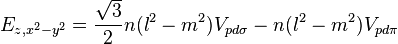
![E_{x,3z^2-r^2} = l[n^2 - (l^2 + m^2)/2]V_{pd\sigma} - \sqrt{3} l n^2 V_{pd\pi}](../I/m/9ecc252f2bda7d0a3e6804c2290ddd44.png)
![E_{y,3z^2-r^2} = m [n^2 - (l^2 + m^2) / 2] V_{pd\sigma} - \sqrt{3} m n^2 V_{pd\pi}](../I/m/80971e2bba73604966bf28418595f404.png)
![E_{z,3z^2-r^2} = n [n^2 - (l^2 + m^2) / 2] V_{pd\sigma} +
\sqrt{3} n (l^2 + m^2) V_{pd\pi}](../I/m/7eba21e36b57f6c89a666d84a8239ea4.png)
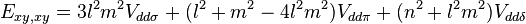
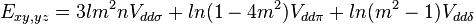
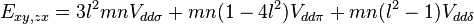
![E_{xy,x^2-y^2} = \frac{3}{2} l m (l^2 - m^2) V_{dd\sigma} +
2 l m (m^2 - l^2) V_{dd\pi} + [l m (l^2 - m^2) / 2] V_{dd\delta}](../I/m/93a1c780ba063b7a401f78963a65e4e3.png)
![E_{yz,x^2-y^2} = \frac{3}{2} m n (l^2 - m^2) V_{dd\sigma} -
m n [1 + 2(l^2 - m^2)] V_{dd\pi} + m n [1 + (l^2 - m^2) / 2] V_{dd\delta}](../I/m/3c905b1cda05070a27e993697b805f05.png)
![E_{zx,x^2-y^2} = \frac{3}{2} n l (l^2 - m^2) V_{dd\sigma} +
n l [1 - 2(l^2 - m^2)] V_{dd\pi} - n l [1 - (l^2 - m^2) / 2] V_{dd\delta}](../I/m/ca950c23558506ab99ae4486dc2c8a70.png)
![E_{xy,3z^2-r^2} = \sqrt{3} \left[ l m (n^2 - (l^2 + m^2) / 2) ]V_{dd\sigma} -
2 l m n^2 V_{dd\pi} + [l m (1 + n^2) / 2] V_{dd\delta} \right]](../I/m/3ac7f43df2765373c9e3543af5255d2b.png)
![E_{yz,3z^2-r^2} = \sqrt{3} \left[ m n (n^2 - (l^2 + m^2) / 2) V_{dd\sigma} +
m n (l^2 + m^2 - n^2) V_{dd\pi} -[ m n (l^2 + m^2) / 2 ]V_{dd\delta} \right]](../I/m/27d2293ff4defdb6163bec27d1755730.png)
![E_{zx,3z^2-r^2} = \sqrt{3} \left[ l n (n^2 - (l^2 + m^2) / 2) V_{dd\sigma} +
l n (l^2 + m^2 - n^2) V_{dd\pi} - [l n (l^2 + m^2) / 2] V_{dd\delta} \right]](../I/m/8878ddfd72999123cc3810d439221d39.png)
![E_{x^2-y^2,x^2-y^2} = \frac{3}{4} (l^2 - m^2)^2 V_{dd\sigma} +
[l^2 + m^2 - (l^2 - m^2)^2] V_{dd\pi} + [n^2 + (l^2 - m^2)^2 / 4] V_{dd\delta}](../I/m/6686f065e4cfe732262118e84b51fac4.png)
![E_{x^2-y^2,3z^2-r^2} = \sqrt{3} \left[
(l^2 - m^2) [n^2 - (l^2 + m^2) / 2] V_{dd\sigma} / 2 + n^2 (m^2 - l^2) V_{dd\pi} +
[(1 + n^2)(l^2 - m^2) / 4 ]V_{dd\delta}\right]](../I/m/0857b9bb721e0c0682f8d944bfddc6f8.png)
![E_{3z^2-r^2,3z^2-r^2} = [n^2 - (l^2 + m^2) / 2]^2 V_{dd\sigma} +
3 n^2 (l^2 + m^2) V_{dd\pi} + \frac{3}{4} (l^2 + m^2)^2 V_{dd\delta}](../I/m/80967c0d44deeb0357416dfd377b1dab.png)
Not all interatomic matrix elements are listed explicitly. Matrix elements that are not listed in this table can be constructed by permutation of indices and cosine directions of other matrix elements in the table.
See also
References
| Wikimedia Commons has media related to Dispersion relations of electrons. |
- 1 2 3 J. C. Slater, G. F. Koster (1954). "Simplified LCAO method for the Periodic Potential Problem". Physical Review 94 (6): 1498–1524. Bibcode:1954PhRv...94.1498S. doi:10.1103/PhysRev.94.1498.
- 1 2 Walter Ashley Harrison (1989). Electronic Structure and the Properties of Solids. Dover Publications. ISBN 0-486-66021-4.
- ↑ As an alternative to neglecting overlap, one may choose as a basis instead of atomic orbitals a set of orbitals based upon atomic orbitals but arranged to be orthogonal to orbitals on other atomic sites, the so-called Löwdin orbitals. See PY Yu & M Cardona (2005). "Tight-binding or LCAO approach to the band structure of semiconductors". Fundamentals of Semiconductors (3 ed.). Springrer. p. 87. ISBN 3-540-25470-6.
- ↑ Orfried Madelung, Introduction to Solid-State Theory (Springer-Verlag, Berlin Heidelberg, 1978).
- ↑ Alexander Altland and Ben Simons (2006). "Interaction effects in the tight-binding system". Condensed Matter Field Theory. Cambridge University Press. pp. 58 ff. ISBN 978-0-521-84508-3.
- ↑ Sir Nevill F Mott & H Jones (1958). "II §4 Motion of electrons in a periodic field". The theory of the properties of metals and alloys (Reprint of Clarendon Press (1936) ed.). Courier Dover Publications. pp. 56 ff. ISBN 0-486-60456-X.
- N. W. Ashcroft and N. D. Mermin, Solid State Physics (Thomson Learning, Toronto, 1976).
- Stephen Blundell Magnetism in Condensed Matter(Oxford, 2001).
- S.Maekawa et al. Physics of Transition Metal Oxides (Spinger-Verlag Berlin Heidelberg, 2004).
- John Singleton Band Theory and Electronic Properties of Solids (Oxford, 2001).
Further reading
- Walter Ashley Harrison (1989). Electronic Structure and the Properties of Solids. Dover Publications. ISBN 0-486-66021-4.
- N. W. Ashcroft and N. D. Mermin (1976). Solid State Physics. Toronto: Thomson Learning.
- Davies, John H. (1998). The physics of low-dimensional semiconductors: An introduction. Cambridge, United Kingdom: Cambridge University Press. ISBN 0-521-48491-X.
- Goringe, C M; Bowler, D R; Hernández, E (1997). "Tight-binding modelling of materials". Reports on Progress in Physics 60 (12): 1447–1512. Bibcode:1997RPPh...60.1447G. doi:10.1088/0034-4885/60/12/001.
- Slater, J. C.; Koster, G. F. (1954). "Simplified LCAO Method for the Periodic Potential Problem". Physical Review 94 (6): 1498–1524. Bibcode:1954PhRv...94.1498S. doi:10.1103/PhysRev.94.1498.
External links
- Crystal-field Theory, Tight-binding Method, and Jahn-Teller Effect in E. Pavarini, E. Koch, F. Anders, and M. Jarrell (eds.): Correlated Electrons: From Models to Materials, Jülich 2012, ISBN 978-3-89336-796-2